Restriction Enzymes: A History
By Wil A.M. Loenen, Leiden University Medical Center
April 2019 · 346 pages, illustrated (38 color and 26 B&W)
ISBN 978-1-621821-05-2
Chapter 4
Chapter doi:10.1101/restrictionenzymes_4
Expansion and Cloning Restriction Enzymes: The 1970s and Early 1980s
EXPANSION AND IDENTIFICATION OF REases IN DIFFERENT BACTERIAL STRAINS
Like others experiencing their “Eureka” or “Aha” moment, Rich Roberts remembers his own “incredible moment” at Harvard in 1972, when he heard a talk by Dan Nathans about specific restriction of SV40 DNA by endonuclease R (HindII; Chapter 3). He immediately realized that REases were the answer to the burning question of how one could generate small DNA fragments that would be ideal for the development of DNA sequencing methods. (Fred Sanger had already developed methods for sequencing RNA, because small molecules such as 5S RNA upon which new approaches could be tested were available. However, no comparably small DNA molecules were available.) After moving to Cold Spring Harbor Laboratory (CSHL) in the autumn of 1972, Rich Roberts entered the R-M field and moved to the forefront right away. In the process, he discovered split genes in adenovirus in 1977 (Chow et al. 1977), for which he would share the Nobel Prize in Physiology or Medicine with Phillip (Phil) Sharp in 1993.
At CSHL, he started by collecting the half dozen known REases: EcoRI and endonuclease R from Ham Smith (Chapter 3), HpaI and HpaII from Phil Sharp (Sharp et al. 1973), and endonuclease Z from Clyde Hutchison III (Middleton et al. 1972). During the purification of endonuclease R, it turned out that the preparation actually contained a second enzyme, and the two enzymes became known as HindII and HindIII following the nomenclature rules first proposed by Smith and Nathans (Smith and Nathans 1973). Likewise, during purification of endonuclease Z it turned out that a second enzyme was present that was called HaeII, and endonuclease Z became renamed to HaeIII. By screening a wide variety of strains from the collection of Jack Strominger that Rich had brought with him to CSHL, 17 new REases were soon identified (including AluI, HhaI, MboI, MboII, and SalI). For the memorable EMBO Workshop (Fig. 1) on “Restriction Enzymes and DNA Sequencing” (organized by Walter Fiers, Jozef [Jeff] Schell, and Marc Van Montagu in Ghent, Belgium in 1974), Rich (Fig. 1A) prepared the first complete REase list, and came up with the name “isoschizomer” to indicate enzymes that recognize the same DNA sequence (Fig. 1B). He expanded and formally published the first comprehensive lists (Roberts 1976a; Roberts 1978), which he would update regularly for many years to come under the name of “The Restriction Enzyme List” (Fig. 2). In the early 1990s, this was stored as a relational database, and in 1993, it was set up as the REBASE database and accompanying website with Dana Macelis (Fig. 3; Roberts and Macelis 1994), which became a valuable resource for the field (Roberts et al. 2015). Luckily, Fred Sanger was also present at the Ghent meeting. He spoke about his novel plus and minus sequencing method (Sanger et al. 1973) and quite happily shared his protocols with Rich, obtaining REases in return.

FIGURE 1. EMBO Workshop “Restriction Enzymes and DNA sequencing” in Ghent, 1974. (A) (Left to right) Ulf Pettersson, Werner Arber, and Rich Roberts at this meeting. (B) Abstract that introduces the word “isoschizomer” to describe enzymes that recognize the same DNA sequence. (Courtesy of Rich Roberts.)

FIGURE 2. Growth of the number of restriction enzymes. From the first NEB catalog. (Courtesy of Rich Roberts.)
FIGURE 3. Homepage of REBASE (http://rebase.neb.com/rebase/rebase.html).
After the Ghent meeting the total number of REases identified rapidly increased to 75 and then >100. The analysis of these was greatly aided by the use of agarose slab gel electrophoresis (Sugden et al. 1975), which rapidly replaced the laborious and slippery vertical tube gels. Slab gels were also used to separate digests of large DNA fragments (e.g., the arms of phage lambda vectors), which had to be run on 0.7% agarose gels for 16–24 h at low voltage to prevent the gel from heating and melting in the middle. Hence, in the laboratory of William (Bill) Brammar in Leicester, the arrival of the submerged slab gel, lovingly called “Concorde” gel after the supersonic jet, was met with great enthusiasm.
The vast majority of the 100-odd REases identified were Type II enzymes, for the simple reason that they gave clear bands on gels and, therefore, analysis was easy. In 1976, Rich published a major overview of all known REases (Roberts 1976b). He tried to get Jim Watson to start a company at CSHL with the profits being used to support research at CSHL, but Jim was not interested because it was considered “unseemly” to be involved with commercial ventures and he also did not think that money could be made from it. Rich then joined forces with Don Comb, who had started a company to sell reagents for research and return the profits back to support research at the company (Biolabs, currently New England Biolabs [NEB]). The first catalog listed 18 purified enzymes and brought in $200,000 within the first year, quite a good return in those days.
Purification of REases was one thing, but getting to know the recognition site was quite another. One had to use 2D electrophoresis and painstakingly analyze the spots. In 1977, the year in which RNA splicing was discovered (Chow et al. 1977), the recognition sequence of BamHI was published in Nature, emphasizing both the difficulty of determining recognition sequences and also illustrating the value of knowing those recognition sequences (Fig. 4; Roberts et al. 1977).
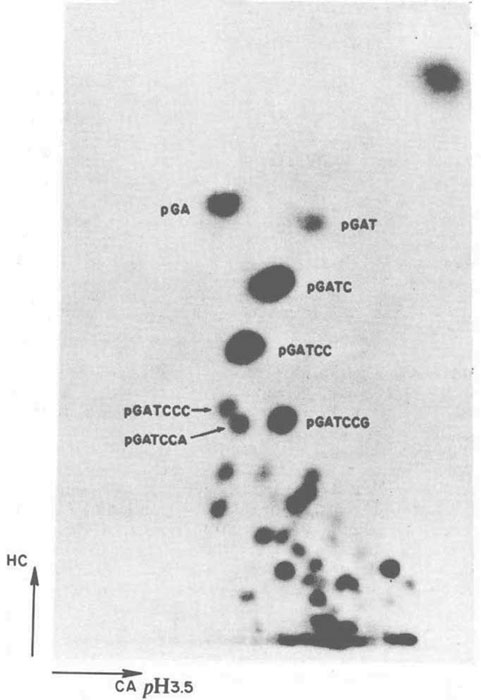
FIGURE 4. Determination of the recognition sequence of BamHI by analysis of radioactive spots after 2D electrophoresis. (Reprinted from Roberts et al. 1977, courtesy of Rich Roberts.)
In the following years, computer programs were developed to aid scientists in finding restriction sites and prepare restriction maps, which were the precursors of the programs currently available, such as REBpredictor (Gingeras et al. 1978), CUTTER, MUTATE, and REMAP (Blumenthal et al. 1982), currently NEBcutter (Vincze et al. 2003).
CLONING OF THE GENES ENCODING REases: THE FIRST R-M SYSTEMS
After the publication of the EcoRI and HindII/III R-M systems, many laboratories tried to clone the genes encoding REases in order to make their purification easier. Others wanted to sequence the systems to see if they could find common motifs. Could one use the corresponding protein sequence to find a common framework for a common DNA specificity? If so, one might be able to generate mutants that would recognize brand-new recognition sequences and save the labor of screening hundreds of new organisms! Despite numerous efforts, this proved impossible because REases are nearly always very different from one another (this is in contrast to the MTases [Pósfai et al. 1989]). Whether this incredible variety and lack of similarity means that they are diverging rapidly from a common ancestor or whether they have evolved independently, remains a matter of debate. It is only relatively recently that a family of restriction enzymes, the Type IIG enzymes, has been shown to be amenable to such engineering, mainly because they have restriction, methylation, and specificity functions in a single polypeptide chain (Morgan and Luyten 2009).
The first R-M system to be cloned was that of Escherichia coli K12 in the laboratories of Noreen Murray and Bill Brammar in Edinburgh in 1976 (Borck et al. 1976). The authors generated an E. coli K12 gene library in phage lambda and selected for recombinant phages that were resistant to EcoKI restriction because of self-modification by M·EcoKI. Some of the other early R-M systems to be identified occurred naturally on small plasmids, making it relatively simple to locate, clone, and sequence the genes and to overproduce the proteins. In addition to EcoRI and EcoRII (Chapter 3) this included EcoRV, PaeR7I, and PvuII (Kosykh et al. 1980; Greene et al. 1981; Newman et al. 1981; Theriault and Roy 1982; Gingeras and Brooks 1983; Bougueleret et al. 1984). The EcoRI and EcoRV REases would be among the first to be crystallized and studied in great detail (Rosenberg et al. 1978; Winkler et al. 1993). In 1978, Ham Smith's group tried to clone the HhaI R-M system from Haemophilus haemolyticus by selection for resistance to phage infection but instead ended up cloning a previously unknown system, which they called HhaII (Mann et al. 1978).
CLONING OF THE GENES ENCODING REases AT NEW ENGLAND BIOLABS, USA1
Around that time Geoffrey (Geoff) Wilson left Noreen Murray's laboratory, where he had cloned the phage T4 gene encoding DNA ligase (Wilson and Murray 1979). He moved to the United States and in 1980 joined NEB, where he started cloning R-M systems with the aim of making overproducing strains for commercial purposes, as well as for fundamental studies. He had his first success by building on the previous work of Walder and colleagues (Walder et al. 1981), successfully making an E. coli strain overexpressing PstI by duplicating the REase gene and expressing it from the strong early lambda promoter pL. The result was a 100-fold increase in the yield of enzyme—and an immediate 10-fold drop in the price NEB charged for it, a win–win result establishing a precedent that continues to this day! In the process of cloning PstI he identified another R-M specificity in Providencia stuartii, named PstII, later shown to be a Type III R-M system (Sears et al. 2005).
Although phage selection led to the successful cloning of the EcoKI (Borck et al. 1976), HhaII (Mann et al. 1978), and PstI (Walder et al. 1981; Loenen 1982) R-M genes, attempts to clone other R-M systems by this means generally failed (e.g., HhaI, HaeII, HaeIII, HpaI, and HpaII). Finding reasons for and solutions to these failures was slow, as new problems were encountered time and time again. Could one perhaps clone enzymes using an in vitro variant of the “methylase-selection” method (Mann et al. 1978)? This procedure, sometimes called the “Hungarian trick” of Pál Venetianer, involved selection for self-modified clones by in vitro DNA restriction and the recovery of survivors by transformation into a nonrestricting host (Szomolanyi et al. 1980). Although this method yielded several cloned MTase genes by 1983, none of these appeared to express the corresponding restriction enzyme or to restrict phage lambda. Did this indicate that some R-M systems might produce too much REase too soon? If so, that would kill their host cell before all of the recognition sites in its DNA had been protectively modified by the incoming MTase. The strategy to solve this “establishment problem” was to clone the MTase gene first, allow full modification of the host DNA to occur, and in a second step acquire the adjacent REase gene. This strategy indeed worked for Joan Brooks at NEB, who used it to clone the DdeI, BamHI, and BglII systems (Howard et al. 1986; Brooks et al. 1989; Anton et al. 1997). However, several R-M systems continued to make little or no restriction enzyme or to restrict phage (e.g., MspI, TaqI, HpaII, and BglI [Walder et al. 1983; Slatko et al. 1987; Nwankwo and Wilson 1988; Lin et al. 1989; Card et al. 1990; Kulakauskas et al. 1994; Newman et al. 1998]). Much later, evidence would emerge for transcriptional regulation of the R genes of a number of Type II enzymes by control (C) proteins and antisense promoters (Tao et al. 1991; Tao and Blumenthal 1992; Liu et al. 2007; Liu and Kobayashi 2007; Mruk et al. 2011).
Quite a different type of cloning problem emerged: Certain E. coli strains do not tolerate m5C methylation and hence prohibit the cloning of this type of MTase genes (Noyer-Weidner et al. 1986; Raleigh and Wilson 1986; Raleigh et al. 1988). The genes involved, named modified cytosine restriction (mcr), would prove to be linked to Luria's paper on restriction of T* mutant phages (by Rgl enzymes: restriction of glucoseless DNA [Luria and Human 1952; Revel and Luria 1970; Noyer-Weidner et al. 1986; Raleigh and Wilson 1986]). And there was more trouble to come: Joe Heitman and Peter Model found another REase activity in E. coli that threw a spanner in the works of cloning people. They discovered that some m6A MTases induced an SOS response in the cell because of DNA damage, which activated yet another enzyme, named Mrr (modified DNA rejection and restriction) (Heitman and Model 1987; see also Halford 2009).
By 1988, Keith Lunnen and others in Geoff Wilson's group had cloned part or all of 38 different R-M systems, and three other laboratories had been set up to do the same by Joan Brooks, Elizabeth (Lise) Raleigh, and Jack Benner (Lunnen et al. 1988; Wilson 1988). That year the first NEB meeting on R-M systems was held in Gloucester, Massachusetts and the proceedings were published in a special issue of Gene, Vol. 74 (Issue 1, 1988). By the time of the CSHL meeting in 2013, NEB scientists had published more than 825 papers, the majority on R-M systems.
CLONING OF THE GENES ENCODING REases IN VILNIUS, LITHUANIA2
NEB was not the only company to look for REases for commercial purposes. At the Institute of Biotechnology (Vilnius, Lithuania), Arvydas Janulaitis started his research and development of REases in 1975 under Soviet rule. He would slowly build an “Enzyme Empire” against all odds (Dickman 1992) and created one of the largest REase collections in the world. From 1985 onward, Arvydas Lubys was involved in this laboratory with the production of optimized tools for molecular biology, including suitable plasmid vectors. Like at NEB, initially they also used phage screening (Mann et al. 1978). The advantage of this procedure was that no purified enzyme was needed for in vitro selection, but unfortunately false positives abounded. They switched to biochemical selection (Szomolanyi et al. 1980), which had the advantage that the library could be screened for multiple MTases in the gene pool.
In 1991, Lithuania became an independent state and the company Fermentas emerged as a spin-off from the Institute of Applied Enzymology. With the aid of EU Structural Fund money, they screened a large number of bacteria for isoschizomers and enzymes of new specificity, purified and evaluated biochemical properties and yields of the REases, cloned R-M systems and sequenced the genes, and developed overexpressing strains and optimal purification protocols. By 1994 they had screened approximately 19,000 bacterial strains; by 2008 this figure had risen to more than 80,000. They implemented ISO standards, producing enzymes in so-called clean rooms, and designed a universal “FastDigest” buffer that enables any combination of restriction enzymes to work simultaneously in one reaction tube allowing full digestion by two (or more) REases at the same temperature in 5 min (Janulaitis 2013), rather than the often laborious and time-consuming traditional way of sequential double digests in different buffers. The combination of biochemical selection and two-step cloning enabled them to clone nearly any R-M system in E. coli devoid of the Mcr and Mrr REases. By the time of the CSHL meeting in 2013, they had cloned and expressed 80 Type IIP, 30 Type IIS, and four Type IIB R-M systems.
In 2003, Rich Roberts and Lise Raleigh visited Fermentas International, Inc. to negotiate product licensing (Fig. 5). In May 2010, all shares of Fermentas International, Inc. were sold to ThermoFisher Scientific.

FIGURE 5. The delegation from NEB in Vilnius to negotiate licensing with Fermentas International, Inc. (Left to right) Arvydas Lubys, Egle Radzeviciene, Algimantas Markauskas (then follow NEB people Elizabeth Raleigh, Rich Roberts, Jurate Bitinaite), Viktoras Butkus, and Arvydas Janulaitis. (Courtesy of Arvydas Janulaitis.)
METEORIC RISE IN THE DISCOVERY OF NEW REases AND THEIR RECOGNITION SEQUENCES
As mentioned earlier, in contrast to the REases, the MTases share identifiable, well-defined common amino acid sequence motifs (Fig. 6). This is independent of whether the MTases flip adenine or cytosine out of the helix for methylation to m6A, m4C, or m5C (Klimasauskas et al. 1989, 1994). These motifs made them highly useful for the discovery of new RM systems in DNA sequences. In 1992, a program called SeqWare, developed by Janos Pósfai, became available at NEB based on this knowledge: SeqWare uses the MTase motifs as a starting point to identify potential R-M systems in GenBank and find the accompanying REases, which are invariably located next, or close, to the MTase genes.
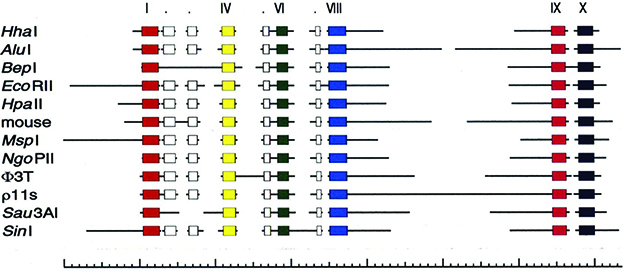
FIGURE 6. Motifs in m5C MTases associated with catalytic methylation (Pósfai et al. 1988, 1989). (Adapted from Kumar et al. 1994; originally adapted from Pósfai et al. 1988, with permission from Elsevier.)
Sanger sequencing simplified, accelerated, and facilitated DNA sequencing for three decades, but in recent years next-generation sequencing from several companies has transformed our ability to determine bacterial and archaeal DNA sequences. Most recently, single-molecule sequencing has become possible (Eid et al. 2009) and importantly can be used to find sites of DNA methylation (Flusberg et al. 2010). This so-called SMRT sequencing method from Pacific Biosciences basically measures how long it takes to add a nucleotide from one position to the next. If on the template a modified base is present, it takes the polymerase a little longer to incorporate across from a methylated base. This shows up as a lag in the profile such that by simply measuring the exact time of the lag, one can work out where the methylated bases are. This works really well for unknown MTases that methylate m6A and m4C but not so well for m5C (Clark et al. 2012). The procedure applies not only to the Type II enzymes but also and most importantly to the Type I and Type III systems that had always been difficult to work with. Type I enzymes cut randomly and give no discrete bands on gels. Type III enzymes rarely give complete cleavage; hence, they give no clean and useful fragmentation patterns. Their specificity can only be deduced by monitoring their methylation patterns. This is the reason that toward the end of the twentieth century, only 20 Type I and 5 Type III enzymes had been identified versus approximately 2400 Type II REases (with more than 200 specificities) (Bickle and Kruger 1993; Roberts and Halford 1993). For the Type I and Type III R-M systems, the motifs and overall sequences of the MTases enable unambiguous identification of their types and the specificity and restriction subunits are usually located close by. It is becoming clear that many bacterial strains have multiple R-M systems—either complete, partial, or mutated. Currently, for every Type I there are roughly two Type II enzymes, although it varies a lot from one strain to the other. How easy is it to find all of them in a whole genome? Can you get that sort of information out of a whole genome? The answer is yes (Clark et al. 2012). In one study, six genomes were analyzed in this way and 27 MTases were found, 13 of which proved to have novel specificities (Murray et al. 2012). By the time of the CSHL meeting in October 2013, data on no less than 360 genomes had been obtained in 18 months.
The era of a brand-new discovery phase had arrived. Although outside the scope of this book, many of these MTases have no corresponding REases. Does this indicate extensive epigenetics in bacteria? If so, is this because in the “wild” bacteria have to adapt rapidly to environmental changes and DNA methylation helps them do so?
REFERENCES
Anton BP, Heiter DF, Benner JS, Hess EJ, Greenough L, Moran LS, Slatko BE, Brooks JE. 1997. Cloning and characterization of the BglII restriction-modification system reveals a possible evolutionary footprint. Gene 187: 19–27. 10.1016/S0378-1119(96)00638-5
Bickle TA, Kruger DH. 1993. Biology of DNA restriction. Microbiol Rev 57: 434–450.
Blumenthal RM, Rice PJ, Roberts RJ. 1982. Computer programs for nucleic acid sequence manipulation. Nucleic Acids Res 10: 91–101. 10.1093/nar/10.1.91
Borck K, Beggs JD, Brammar WJ, Hopkins AS, Murray NE. 1976. The construction in vitro of transducing derivatives of phage lambda. Mol Gen Genet 146: 199–207. 10.1007/BF00268089
Bougueleret L, Schwarzstein M, Tsugita A, Zabeau M. 1984. Characterization of the genes coding for the Eco RV restriction and modification system of Escherichia coli. Nucleic Acids Res 12: 3659–3676. 10.1093/nar/12.8.3659
Brooks JE, Benner JS, Heiter DF, Silber KR, Sznyter LA, Jager-Quinton T, Moran LS, Slatko BE, Wilson GG, Nwankwo DO. 1989. Cloning the BamHI restriction modification system. Nucleic Acids Res 17: 979–997. 10.1093/nar/17.3.979
Card CO, Wilson GG, Weule K, Hasapes J, Kiss A, Roberts RJ. 1990. Cloning and characterization of the HpaII methylase gene. Nucleic Acids Res 18: 1377–1383. 10.1093/nar/18.6.1377
Chow LT, Gelinas RE, Broker TR, Roberts RJ. 1977. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12: 1–8. 10.1016/0092-8674(77)90180-5
Clark TA, Murray IA, Morgan RD, Kislyuk AO, Spittle KE, Boitano M, Fomenkov A, Roberts RJ, Korlach J. 2012. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res 40: e29. 10.1093/nar/gkr1146
Dickman S. 1992. Lithuanian biochemist builds enzyme empire. Science 257: 1473–1474. 10.1126/science.1326121
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138. 10.1126/science.1162986
Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Meth 7: 461–465. 10.1038/nmeth.1459
Gingeras TR, Brooks JE. 1983. Cloned restriction/modification system from Pseudomonas aeruginosa. Proc Natl Acad Sci 80: 402–406. 10.1073/pnas.80.2.402
Gingeras TR, Milazzo JP, Roberts RJ. 1978. A computer assisted method for the determination of restriction enzyme recognition sites. Nucleic Acids Res 5: 4105–4127. 10.1093/nar/5.11.4105
Greene PJ, Gupta M, Boyer HW, Brown WE, Rosenberg JM. 1981. Sequence analysis of the DNA encoding the Eco RI endonuclease and methylase. J Biol Chem 256: 2143–2153.
Halford SE. 2009. The (billion dollar) consequences of studying why certain isolates of phage lambda infect only certain strains of E. coli: restriction enzymes. Biochemist 31: 10–13.
Heitman J, Model P. 1987. Site-specific methylases induce the SOS DNA repair response in Escherichia coli. J Bacteriol 169: 3243–3250. 10.1128/jb.169.7.3243-3250.1987
Howard KA, Card C, Benner JS, Callahan HL, Maunus R, Silber K, Wilson G, Brooks JE. 1986. Cloning the DdeI restriction-modification system using a two-step method. Nucleic Acids Res 14: 7939–7951. 10.1093/nar/14.20.7939
Janulaitis A. 2013. Cut and go—FastDigest with all restriction enzymes at same temperature and buffer: a new paradigm in DNA digestion. In Modern biopharmaceuticals: recent success stories (ed. Knäblein J), pp. 135–146. Wiley-VCH, Weinheim.
Klimasauskas S, Timinskas A, Menkevicius S, Butkiene D, Butkus V, Janulaitis A. 1989. Sequence motifs characteristic of DNA[cytosine-N4]methyltransferases: similarity to adenine and cytosine-C5 DNA-methylases. Nucleic Acids Res 17: 9823–9832. 10.1093/nar/17.23.9823
Klimasauskas S, Kumar S, Roberts RJ, Cheng X. 1994. HhaI methyltransferase flips its target base out of the DNA helix. Cell 76: 357–369. 10.1016/0092-8674(94)90342-5
Kosykh VG, Buryanov YI, Bayev AA. 1980. Molecular cloning of EcoRII endonuclease and methylase genes. Mol Gen Genet 178: 717–718. 10.1007/BF00337884
Kulakauskas S, Barsomian JM, Lubys A, Roberts RJ, Wilson GG. 1994. Organization and sequence of the HpaII restriction-modification system and adjacent genes. Gene 142: 9–15. 10.1016/0378-1119(94)90348-4
Kumar S, Cheng X, Klimasauskas S, Mi S, Pósfai J, Roberts RJ, Wilson GG. 1994. The DNA (cytosine-5) methyltransferases. Nucleic Acids Res 22: 1–10. 10.1093/nar/22.1.1
Lin PM, Lee CH, Roberts RJ. 1989. Cloning and characterization of the genes encoding the MspI restriction modification system. Nucleic Acids Res 17: 3001–3011. 10.1093/nar/17.8.3001
Liu Y, Kobayashi I. 2007. Negative regulation of the EcoRI restriction enzyme gene is associated with intragenic reverse promoters. J Bacteriol 189: 6928–6935. 10.1128/JB.00127-07
Liu Y, Ichige A, Kobayashi I. 2007. Regulation of the EcoRI restriction-modification system: identification of ecoRIM gene promoters and their upstream negative regulators in the ecoRIR gene. Gene 400: 140–149. 10.1016/j.gene.2007.06.006
Loenen WAM. 1982. “The construction and use of lambda vectors in molecular cloning.” PhD thesis, University of Leicester.
Lunnen KD, Barsomian JM, Camp RR, Card CO, Chen SZ, Croft R, Looney MC, Meda MM, Moran LS, Nwankwo DO, et al. 1988. Cloning type-II restriction and modification genes. Gene 74: 25–32. 10.1016/0378-1119(88)90242-9
Luria SE, Human ML. 1952. A nonhereditary, host-induced variation of bacterial viruses. J Bacteriol 64: 557–569.
Mann MB, Rao RN, Smith HO. 1978. Cloning of restriction and modification genes in E. coli: the HhaII system from Haemophilus haemolyticus. Gene 3: 97–112. 10.1016/0378-1119(78)90054-9
Middleton JH, Edgell MH, Hutchison CA 3rd. 1972. Specific fragments of ϕX174 deoxyribonucleic acid produced by a restriction enzyme from Haemophilus aegyptius, endonuclease Z. J Virol 10: 42–50.
Morgan RD, Luyten YA. 2009. Rational engineering of type II restriction endonuclease DNA binding and cleavage specificity. Nucleic Acids Res 37: 5222–5233. 10.1093/nar/gkp535
Mruk I, Liu Y, Ge L, Kobayashi I. 2011. Antisense RNA associated with biological regulation of a restriction-modification system. Nucleic Acids Res 39: 5622–5632. 10.1093/nar/gkr166
Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. 2012. The methylomes of six bacteria. Nucleic Acids Res 40: 11450–11462. 10.1093/nar/gks891
Newman AK, Rubin RA, Kim SH, Modrich P. 1981. DNA sequences of structural genes for Eco RI DNA restriction and modification enzymes. J Biol Chem 256: 2131–2139.
Newman M, Lunnen K, Wilson G, Greci J, Schildkraut I, Phillips SE. 1998. Crystal structure of restriction endonuclease BglI bound to its interrupted DNA recognition sequence. EMBO J 17: 5466–5476. 10.1093/emboj/17.18.5466
Noyer-Weidner M, Diaz R, Reiners L. 1986. Cytosine-specific DNA modification interferes with plasmid establishment in Escherichia coli K12: involvement of rglB. Mol Gen Genet 205: 469–475. 10.1007/BF00338084
Nwankwo DO, Wilson GG. 1988. Cloning and expression of the MspI restriction and modification genes. Gene 64: 1–8. 10.1016/0378-1119(88)90475-1
Pósfai J, Bhagwat AS, Roberts RJ. 1988. Sequence motifs specific for cytosine methyltransferases. Gene 74: 261–265. 10.1016/0378-1119(88)90299-5
Pósfai J, Bhagwat AS, Pósfai G, Roberts RJ. 1989. Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res 17: 2421–2435. 10.1093/nar/17.7.2421
Raleigh EA, Wilson G. 1986. Escherichia coli K-12 restricts DNA containing 5-methylcytosine. Proc Natl Acad Sci 83: 9070–9074. 10.1073/pnas.83.23.9070
Raleigh EA, Murray NE, Revel H, Blumenthal RM, Westaway D, Reith AD, Rigby PW, Elhai J, Hanahan D. 1988. McrA and McrB restriction phenotypes of some E. coli strains and implications for gene cloning. Nucleic Acids Res 16: 1563–1575. 10.1093/nar/16.4.1563
Revel HR, Luria SE. 1970. DNA-glucosylation in T-even phage: genetic determination and role in phage-host interaction. Ann Rev Genet 4: 177–192. 10.1146/annurev.ge.04.120170.001141
Roberts RJ. 1976a. Restriction and modification enzymes and their recognition sequences. In CRC handbook of biochemistry and molecular biology, 3rd ed., Nucleic Acids Vol II, pp. 532–535. CRC Press, Boca Raton, FL.
Roberts RJ. 1976b. Restriction endonucleases. CRC Crit Rev Biochem 4: 123–164. 10.3109/10409237609105456
Roberts RJ. 1978. Restriction and modification enzymes and their recognition sequences. Gene 4: 183–194. 10.1016/0378-1119(78)90017-3
Roberts RJ, Halford SE. 1993. Type II restriction enzymes. In Nucleases, 2nd ed. (ed. Linn SM, et al. ), pp. 35–88, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Roberts RJ, Macelis D. 1994. REBASE—restriction enzymes and methylases. Nucleic Acids Res 22: 3628–3639. 10.1093/nar/22.17.3628
Roberts RJ, Wilson GA, Young FE. 1977. Recognition sequence of specific endonuclease BamH.I from Bacillus amyloliquefaciens H. Nature 265: 82–84. 10.1038/265082a0
Roberts RJ, Vincze T, Pósfai J, Macelis D. 2015. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43: D298–D299. 10.1093/nar/gku1046
Rosenberg JM, Dickerson RE, Greene PJ, Boyer HW. 1978. Preliminary X-ray diffraction analysis of crystalline EcoRI endonuclease. J Mol Biol 122: 241–245. 10.1016/0022-2836(78)90039-6
Sanger F, Donelson JE, Coulson AR, Kossel H, Fischer D. 1973. Use of DNA polymerase I primed by a synthetic oligonucleotide to determine a nucleotide sequence in phage fl DNA. Proc Natl Acad Sci 70: 1209–1213. 10.1073/pnas.70.4.1209
Sears A, Peakman LJ, Wilson GG, Szczelkun MD. 2005. Characterization of the Type III restriction endonuclease PstII from Providencia stuartii. Nucleic Acids Res 33: 4775–4787. 10.1093/nar/gki787
Sharp PA, Sugden B, Sambrook J. 1973. Detection of two restriction endonuclease activities in Haemophilus parainfluenzae using analytical agarose–ethidium bromide electrophoresis. Biochemistry 12: 3055–3063. 10.1021/bi00740a018
Slatko BE, Benner JS, Jager-Quinton T, Moran LS, Simcox TG, Van Cott EM, Wilson GG. 1987. Cloning, sequencing and expression of the Taq I restriction-modification system. Nucleic Acids Res 15: 9781–9796. 10.1093/nar/15.23.9781
Smith HO, Nathans D. 1973. Letter: a suggested nomenclature for bacterial host modification and restriction systems and their enzymes. J Mol Biol 81: 419–423. 10.1016/0022-2836(73)90152-6
Sugden B, De Troy B, Roberts RJ, Sambrook J. 1975. Agarose slab-gel electrophoresis equipment. Anal Biochem 68: 36–46. 10.1016/0003-2697(75)90676-4
Szomolanyi E, Kiss A, Venetianer P. 1980. Cloning the modification methylase gene of Bacillus sphaericus R in Escherichia coli. Gene 10: 219–225. 10.1016/0378-1119(80)90051-7
Tao T, Blumenthal RM. 1992. Sequence and characterization of pvuIIR, the PvuII endonuclease gene, and of pvuIIC, its regulatory gene. J Bacteriol 174: 3395–3398. 10.1128/jb.174.10.3395-3398.1992
Tao T, Bourne JC, Blumenthal RM. 1991. A family of regulatory genes associated with type II restriction-modification systems. J Bacteriol 173: 1367–1375. 10.1128/jb.173.4.1367-1375.1991
Theriault G, Roy PH. 1982. Cloning of Pseudomonas plasmid pMG7 and its restriction-modification system in Escherichia coli. Gene 19: 355–359. 10.1016/0378-1119(82)90026-9
Vincze T, Pósfai J, Roberts RJ. 2003. NEBcutter: a program to cleave DNA with restriction enzymes. Nucleic Acids Res 31: 3688–3691. 10.1093/nar/gkg526
Walder RY, Hartley JL, Donelson JE, Walder JA. 1981. Cloning and expression of the Pst I restriction-modification system in Escherichia coli. Proc Natl Acad Sci 78: 1503–1507. 10.1073/pnas.78.3.1503
Walder RY, Langtimm CJ, Chatterjee R, Walder JA. 1983. Cloning of the MspI modification enzyme. The site of modification and its effects on cleavage by MspI and HpaII. J Biol Chem 258: 1235–1241.
Wilson GG. 1988. Cloned restriction-modification systems—a review. Gene 74: 281–289. 10.1016/0378-1119(88)90304-6
Wilson GG, Murray NE. 1979. Molecular cloning of the DNA ligase gene from bacteriophage T4. I. Characterisation of the recombinants. J Mol Biol 132: 471–491. 10.1016/0022-2836(79)90270-5
Winkler FK, Banner DW, Oefner C, Tsernoglou D, Brown RS, Heathman SP, Bryan RK, Martin PD, Petratos K, Wilson KS. 1993. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J 12: 1781–1795. 10.1002/j.1460-2075.1993.tb05826.x
WWW RESOURCES
http://library.cshl.edu/Meetings/restriction-enzymes/v-GeoffWilson.php Wilson G. 2013. The cloning efforts at NEB.
http://library.cshl.edu/Meetings/restriction-enzymes/v-Janulaitis.php Janulaitis A. 2013. Science and politics: three phases of commercialization at Fermentas.
http://library.cshl.edu/Meetings/restriction-enzymes/v-Lubys.php Lubys A. 2013. The cloning efforts at Fermentas.
http://library.cshl.edu/Meetings/restriction-enzymes/v-Roberts.php Roberts R. 2013. Many more REs at CSHL, the start of REBASE and more recent work.
http://rebase.neb.com/rebase/rebase.html The Restriction Enzyme Database.
1 http://library.cshl.edu/Meetings/restriction-enzymes/v-GeoffWilson.php; http://library.cshl.edu/Meetings/restriction-enzymes/v-Roberts.php.
2 http://library.cshl.edu/Meetings/restriction-enzymes/v-Janulaitis.php; http://library.cshl.edu/Meetings/restriction-enzymes/v-Lubys.php.
