Restriction Enzymes: A History
By Wil A.M. Loenen, Leiden University Medical Center
April 2019 · 346 pages, illustrated (38 color and 26 B&W)
ISBN 978-1-621821-05-2
Chapter 3
Chapter doi:10.1101/restrictionenzymes_3
The Discovery of Type II Restriction Enzymes: The 1970s
THE DISCOVERY OF HindII IN THE LABORATORY OF HAMILTON SMITH1
As described in the previous chapters, early work on R-M enzymes had focused on the enzymes of Escherichia coli K12 and B and phage P1. The work by the Geneva group of Werner Arber supported the notion that the enzymes would recognize a specific sequence, but the determination of this sequence was no easy matter. As chance brought Werner Arber into the restriction field, it was also by chance that Hamilton (Ham) Smith at Johns Hopkins identified the first recognition site of a REase that belongs to a family of enzymes that would change the landscape of molecular biology. Trained as a clinician, Ham Smith's vocation in life had changed some years earlier when reading the Watson and Crick paper on the model of the DNA double helix (Watson and Crick 1953). In a journal club he discussed another interesting paper, that of the purification and in vitro study of the EcoKI enzyme (Chapter 2; Meselson and Yuan 1968). Kent Wilcox from his laboratory was present at this seminar. When a little later he encountered rapid degradation of phage 32P-labeled P22 DNA after transformation in competent cells of Haemophilus influenza, Kent wondered: Was the degradation of P22 DNA perhaps also due to restriction? Although skeptical at first, that night at home Ham Smith realized that this could be tested in the Ostwald viscometer (Fig. 1) (http://library.cshl.edu/Meetings/restriction-enzymes/v-Smith.php. 2013). Changes in the viscosity of the DNA would relate to changes in the size of that DNA!
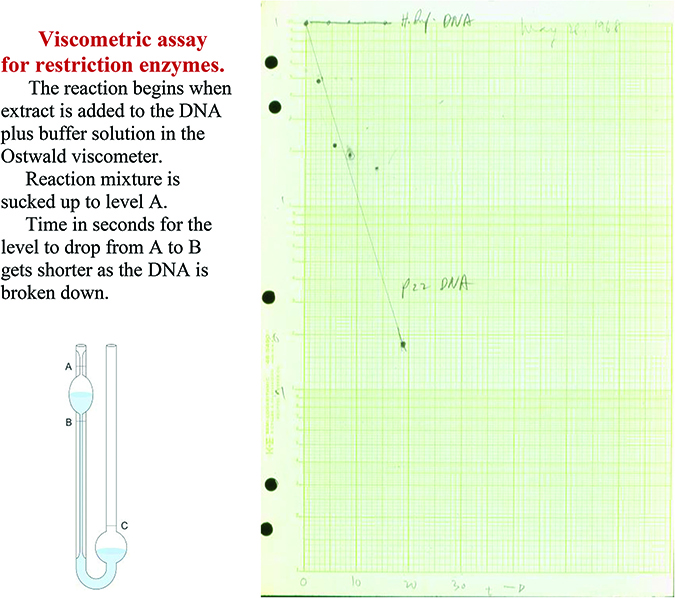
FIGURE 1. The viscosity meter experiment, May 1968, to measure degradation of foreign P22 DNA in H. influenza. (Courtesy of Ham Smith.)
And indeed, in the presence of Mg2+ the viscosity of the P22 DNA dropped rapidly within 2 minutes after addition of the cell extract, whereas that of the control H. influenzae DNA remained constant. The rate of this degradation of the “foreign” P22 DNA was proportional to the time (Smith and Wilcox 1970). Purification of this activity confirmed the existence in the extract of a nuclease that recognized the foreign DNA but not the bacterial DNA. This enzyme, called EndoR, later renamed HindII, not only cleaved DNA of P22, but also that of phage T7 (±70 times). Alkaline gradients (that separate the DNA strands) showed the cuts to be double-strand breaks. Did this enzyme cut a specific sequence, and if so, could one sequence the actual end(s)?
Ham Smith recalls their luck in having Bernard (Bernie) Weiss on the same floor of their building (Fig. 2, left). Bernie Weiss had come from Charles (Charlie) Richardson's laboratory at Harvard (where he had discovered T4 DNA ligase). Bernie Weiss supplied them with T4 polynucleotide kinase (pnk) and homemade γ-32P ATP “that was so hot that it turned brown from radiation” (http://library.cshl.edu/Meetings/restriction-enzymes/v-Smith.php. 2013). Labeling the 5′ termini of the cleavage products with pnk allowed digestion of the DNA with various nucleases to obtain labeled oligonucleotides, which were analyzed by chromatography and electrophoresis. These procedures revealed the dinucleotide to be AA and GA. Ham Smith gave a talk about this at the Federation meeting in Atlantic City in April 1969 and recalls that there was little general interest in their results, although Stu Linn recalls that he was very much interested indeed.

FIGURE 2. (Left) Members of the Department of Microbiology at Johns Hopkins in the mid-1970s. (From left to right) Ken Berns, Tom Kelly, Dan Nathans, Ham Smith, and Bernie Weiss. (Right) Kathleen Danna and Dan Nathans at the CSH Tumor Virus Meeting, summer 1970. (Courtesy of Ham Smith.)
But what was the nature of the structure? When Tom Kelly arrived as a postdoc, he could use newly commercially available 33P in combination with 5′ 32P label and thin-layer electrophoresis. This allowed him to prove the specific recognition sequence to consist of six bases. There were three possibilities: the breaks could be opposite each other (Fig. 3A) or away from each other with either a 5′ or 3′ overhang (Fig. 3B or 3C).
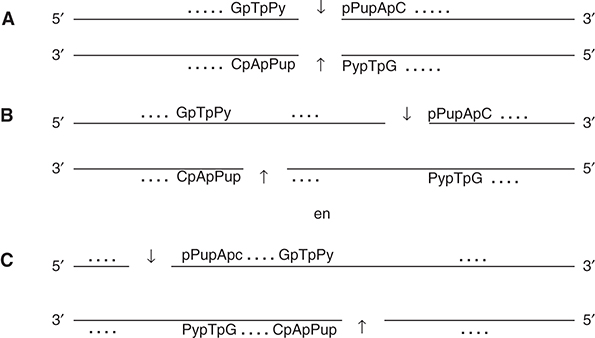
FIGURE 3. Possible structures of the EndoR/HindII recognition site. The cuts on the two DNA strands could be blunt end (A) or staggered with either a 3′ or 5′ overhang (B and C).
Subsequent experiments showed the cut to be blunt end with no intervening bases; hence, structure A in Figure 3 was correct. The recognition sequence of EndoR/HindII 5′-GTPy-PuAC-3′ was published in 1970, back-to-back with a paper on the purification of the enzyme (Kelly and Smith 1970; Smith and Wilcox 1970). In contrast to the lack of interest the year before at the Federation meeting, this time the impact was enormous and Salva Luria and Werner Arber sent their congratulations.
Around that time, Dan Nathans, who also worked on the same floor as Ham Smith, was about to return from sabbatical leave in Israel, where he had learned to handle the oncogenic simian virus SV40. He felt that the cancer field was about to explode and he wanted to look at transformation by this eukaryotic virus. Could he test EndoR on SV40 DNA? Initially he and PhD student Kathleen Danna (Fig. 2, right) used sucrose gradients and polyacrylamide tube gels, which failed to give clean and clear results. When they resorted to radiography of dried-down gels, however, resolution was excellent. In the absence of DNA markers, Kathleen Danna mapped the sites on the SV40 DNA using hundreds of gels with partial digests: partials had to be isolated, digested again with the same or another enzyme, ordered, and the sizes determined with the help of the label, a tedious procedure (Danna and Nathans 1971). In 1973 they published the first map of SV40 (Danna et al. 1973). Dan Nathans then proposed a nomenclature for R-M systems (Smith and Nathans 1973): a three-letter abbreviation for the host strain plus a subtype designation, where necessary (e.g., EcoK for E. coli K12 or Hind for H. influenza strain d). Different R-M systems in a strain would be indicated by roman numerals (e.g., EcoKI).
Together with Werner Arber and Ham Smith, Dan Nathans was awarded the Nobel Prize in Physiology or Medicine in 1978. Sadly, he passed away in 1999 (Danna 2010).
THE DISCOVERY OF EcoRI
One of the best-known enzymes discovered in the early 1970s was identified in the laboratory of Herb Boyer at University of California, San Francisco (UCSF). Herb Boyer developed a strong interest in genetics and chemistry fueled by his science and math teacher Pat Bucci and was “completely blown away by the structure and heuristic value of the structure” of the model of the DNA double helix (http://library.cshl.edu/Meetings/restriction-enzymes/v-Boyer.php. 2013). How he entered the restriction field will sound familiar: For his PhD thesis he had to analyze the regulation of the arabinose operon using conjugation experiments between E. coli K12 and E. coli B strains. Thus he encountered host-controlled variation. After his move to UCSF, he asked Daisy Roulland-Dussoix to purify the E. coli B enzyme, EcoBI, which she did (Chapter 2). But when Stu Linn told them that EcoBI cut randomly, the project was abandoned.
In the meantime, Herb Boyer had come across the pioneering papers by Tsutomu Watanabe and coworkers in Tokyo on multidrug resistance in clinical strains, a major health hazard in Japan in the early 1950s (Watanabe et al. 1964, 1966; Takano et al. 1966, 1968). Apparently, drug resistance was located on extrachromosomal DNA, and, in addition, some of these episomes also showed nuclease activity. They called these resistance transfer factors (RTFs). During the analysis of these RTFs, they were the first to provide evidence for restriction of lambda DNA in vitro, which required Mg2+ for activity (Watanabe et al. 1964; Takano et al. 1966). Herb Boyer wondered: Would clinical strains collected in California also carry such restricting RTFs? He asked PhD student Bob Yoshimori to identify such restricting RTF factors in the hospital collection. He should try to test this by transfer of the RTF plasmids from the clinical backgrounds into E. coli K12. This search resulted in two different restriction specificities: one was the same as one of Watanabe's RTFs, named EcoRII, but the other one had a novel specificity, called EcoRI. Like HindII, and in contrast to EcoKI and EcoBI, both required only Mg2+ for activity. Moreover, the recognition site of EcoRI (GAATTC) did not have an ambiguity like the HindII site, and to their surprise and delight, the enzyme cut after the first G on both strands producing “AATT” sticky ends (Yoshimori et al. 1972).
Could the symmetrical sticky ends make it possible to mix fragments of different DNAs and transform them into E. coli? The answer to that was positive: Genetic engineering was born. EcoRI would become one of the most important enzymes of the decade, as it made a limited number of cuts on lambda DNA and various plasmids and a single one on SV40 DNA (Hedgpeth et al. 1972; Jackson et al. 1972; Cohen et al. 1973). This led to the construction of cloning vectors based on lambda, plasmids, and SV40. In these constructions the new protocol for the analysis of DNA fragments using agarose gels combined with staining with ethidium bromide greatly simplified the analysis of both size and number of fragments made by the enzymes (Sharp et al. 1973).
THE CLASSIFICATION OF THE RESTRICTION-MODIFICATION ENZYMES INTO FOUR MAIN TYPES
The differences between the enzymes encoded by E. coli, H. influenza, and the RTF plasmids led to the classification in S-adenosylmethionine (SAM)-dependent Type I and SAM-independent Type II enzymes (Boyer 1971). A little later, new evidence warranted a further division of the SAM-dependent enzymes: Methionine analogs (such as ethionine, which in contrast to methionine does allow protein synthesis to proceed) revealed two classes of enzymes: those truly dependent on SAM for restriction (Type I enzymes: no REase activity in the absence of SAM → stable DNA in the presence of analogs → survival of the cells), and those that were less dependent on SAM (REase active → cells self-destruct in the presence of analogs). The latter type, which includes the phage P1 enzyme, was renamed Type III in 1978 (Kauc and Piekarowicz 1978).
The story does not end there, as Luria's T* mutant phages (Chapter 1) led to the discovery of another class of restriction enzymes. T* mutants are unable to add glucose residues to the hydroxymethylcytosine (hm5C) bases in their DNA. hm5C blocks a great many restriction enzymes, but not the E. coli Rgl1 and Rgl2 (restriction of
The methylation- and/or other modification-dependent enzymes caused a lot of cloning trouble when people tried to clone often considerably methylated, eukaryotic DNA in E. coli strains (Raleigh et al. 1988). This led to the discovery that Rgl restricts both m5C and hm5C, which also meant cloning trouble when trying to (over)express some modification enzymes in E. coli. The enzymes were renamed Mcr (methyl cytosine restriction) and classified as Type IV in a multi-authored review in 2003 (Roberts et al. 2003). This review details the characteristics of different subclasses of the Type I and Type II enzymes (see http://rebase.neb.com/rebase/rebase.html for current details).
SUBDIVISIONS
The original definition of Type II enzymes was that they recognize specific DNA sequences and cleave at a constant position at or close to that sequence. As a rule they require only Mg2+ as cofactor, though more and more exceptions to this rule were and are being identified. The Type II enzymes are highly heterogeneous though the subtypes sometimes overlap, and an enzyme may belong to more than one subtype (Roberts et al. 2003). For example, BcgI is both a Type IIB enzyme (cuts on both sites of its recognition sequence) and Type IIC enzyme (DNA cleavage and modification are carried out by one and the same polypeptide). EcoRI is an enzyme that fits the original definition, and this subtype is called Type IIP.
Tom Bickle compared the heterogeneity with “Cuvier's pachyderms” (http://library.cshl.edu/Meetings/restriction-enzymes/v-Bickle.php). Cuvier was a French zoologist who tried to classify animals above species level and grouped animals with thick skins such as rhinos and hippos (called pachyderms in Greek). In this way, organisms (and by extension proteins) that are not genetically and/or evolutionarily linked are classified based on a single particular trait. In the case of REases, Type II enzymes do not (usually) require ATP and/or SAM, which sets them apart from the Type I and Type III enzymes. Similarly, their general inability to cut the recognition site when modified sets them apart from the Type IV enzymes, although some Type II enzymes (such as Type IIM DpnI) will only cut methylated DNA. In evolutionary terms, the Type II enzymes are very highly heterogeneous, both in protein sequences and reaction mechanisms.
SUBSEQUENT ENZYME DISCOVERIES
After the discovery of HindII, EcoRI, EcoRII, and their recognition sequences, several other enzymes soon followed. By 1974, recognition sequences of five additional enzymes (HaeIII, HindIII, HpaI, HpaII, and AvaI) were known, as well as nine other enzymes that recognize the same sequence, so-called “isoschizomers,” and 16 less well-characterized REases (Nathans and Smith 1975). All known sites were 4- to 6-bp-long with twofold rotational symmetry. This led to the idea that the REase would also have twofold symmetry, which would be in line with EcoRI having two identical subunits. Cleavage could be blunt end, like HindII, or staggered, like EcoRI. The enzymes could have a 5′ overhang with 2, 3, 4, or 5 nucleotides, or a 3′ overhang of 2 or 4 nucleotides, posing an interesting mechanistic problem to protein chemists for years to come. In addition to these enzymes cutting at the site, the first example of a Type II enzyme that cuts away from the site was identified with the help of known lambda DNA sequences (Kleid et al. 1976).
How fast the field was expanding becomes clear from the next major survey of the field a little later (Roberts 1976). By 1976, 86 REases were known with 45 different specificities, 22 of these identified in Haemophilus species. The finding of an enzyme in the Gram-positive Bacillus subtilis raised expectations that REases might not be limited to Gram-negative bacteria but might be widespread throughout the bacterial kingdom (Bron et al. 1975). Several facts were puzzling: Why would one strain make a lot of enzyme, whereas another one such a tiny amount that it was impossible to characterize properly? What was the pattern or origin of the REases? They were found on plasmids, phages, (defective) prophages, and on the chromosome. Richard (Rich) Roberts wondered whether perhaps REases were coded by these mobile DNA elements, which would use them “to shuffle genetic information and create new recombinant genomes in the same way that has recently been accomplished in vitro” (Roberts 1976). Did REases exist in higher organisms such as the lower eukaryotes yeast, fungi, or ciliates? Nothing emerged, but such enzymes might be missed because of low amounts, instability, or peculiar cofactors.
EARLY USES OF TYPE II REases IN RECOMBINANT DNA
This topic has been extensively covered in reviews and books, and here a short overview is given of the birth of genetic engineering.
Mapping
One of the first applications of REases was physical mapping of chromosomes (see Nathans and Smith 1975 for details). Specific restriction sites could be correlated with the genetic map and serve as physical reference points. In this way, genes, as well as start and stop signs for transcription and translation, could be mapped. Direction of transcription, origin of DNA replication, and direction of replication could be resolved (Nathans and Smith 1975). Binding sites for enzymes could be located on specific DNA fragments, and the map could also be used to find deletions, rearrangements, insertions, or substitutions that previously relied on EM visualization. Such research could reveal differences between strains of viruses and their evolutionary changes or show recombination events (Nathans and Smith 1975). One interesting early example was the discovery of maternal inheritance of mitochondria: The mule and the hinny never have the same mitochondrial DNA, as this is always inherited from the mother horse or mother donkey and never from the father (Hutchison et al. 1974).
Sequencing
Another application of REases was to simplify nucleotide sequencing (Salser 1974) by providing small DNA restriction fragments from <20 to ±1000 bp. These would be generated by sequential enzyme digestion, starting with enzymes that cleave once or only a few times in a given genome (such as that of SV40, adenovirus, or phage lambda), followed by other enzymes producing overlapping smaller fragments amenable to sequencing. Around this time, novel DNA sequencing protocols (Sanger and Coulson 1975; Maxam and Gilbert 1977) and the useful “Southern blot” technique were published (Southern 1975a).
Isolation of Genes and Analysis of Repeat Sequences
In addition, one could isolate genes (Brown and Stern 1974) or analyze repetitive DNA sequences that gave discrete bands superimposed on the large and long smear of a million or more bands, generated when cutting a mammalian genome (Mowbray and Landy 1974). For example, EcoRII cleaves purified mouse satellite DNA into some 20-odd regularly spaced bands, which are multiples of a 240-bp repeat (Southern 1975b). Methods were not available yet to undertake “detailed analysis of the unique fraction of eukaryotic DNA [which] will require amplification of these sequences artificially, e.g. by molecular cloning. Such DNA segments might then be amenable to the same type of analyses applied to viral chromosomes and satellite DNA” (Nathans and Smith 1975). With the then available techniques, it became easier to analyze repetitive DNA in eukaryotes—satellite repeats, as well as the multiple copies of histone genes and rRNA genes.
Recombinant DNA
One of the most far-reaching applications of REases was the in vitro construction of DNA molecules with novel biological activities. Insertion or deletion mutants or artificial recombinants could be made by joining DNA fragments via the sticky ends (made at the cut site by the REase) or by homopolymer tailing with oligo(dT). Excision, addition, and rearrangement of fragments in a given genome or combination of DNA from various sources into recombinant molecules became possible (Jackson et al. 1972; Cohen and Chang 1973; Cohen et al. 1973; Lobban and Kaiser 1973; Hershfield et al. 1974; Lai and Nathans 1974; Murray and Murray 1974). Such recombined molecules could be cloned in suitable cells and propagated to yield large quantities of the new DNA, and, in some cases, specific gene products (e.g., X. laevis rDNA in pSC101 [Morrow et al. 1974] or trp in lambda [Murray and Murray 1974]). In this way, one also could produce specific transducing viruses for animal cells (Brockman and Nathans 1974; Nathans et al. 1974).
All these exciting new uses of REases in molecular biology, to analyze the DNA of many different organisms, made many forget the biological properties of the REases. A few laboratories did not, and currently the world of REases and their MTases is at least as exciting as the field of genetic engineering and DNA sequencing, as detailed later.
See, for some further background reading, Judson (1979) and Watson and Tooze (1981).
REFERENCES
Boyer HW. 1971. DNA restriction and modification mechanisms in bacteria. Ann Rev Microbiol 25: 153–176. 10.1146/annurev.mi.25.100171.001101
Brockman WW, Nathans D. 1974. The isolation of simian virus 40 variants with specifically altered genomes. Proc Natl Acad Sci 71: 942–946. 10.1073/pnas.71.3.942
Bron S, Murray K, Trautner TA. 1975. Restriction and modification in B. subtilis. Purification and general properties of a restriction endonuclease from strain R. Mol Gen Genet 143: 13–23. 10.1007/BF00269416
Brown DD, Stern R. 1974. Methods of gene isolation. Ann Rev Biochem 43: 667–693. 10.1146/annurev.bi.43.070174.003315
Cohen SN, Chang AC. 1973. Recircularization and autonomous replication of a sheared R-factor DNA segment in Escherichia coli transformants. Proc Natl Acad Sci 70: 1293–1297. 10.1073/pnas.70.5.1293
Cohen SN, Chang AC, Boyer HW, Helling RB. 1973. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci 70: 3240–3244. 10.1073/pnas.70.11.3240
Danna D. 2010. Biographical memoir. Proc Am Philos Soc 154: 338–354.
Danna K, Nathans D. 1971. Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc Natl Acad Sci 68: 2913–2917. 10.1073/pnas.68.12.2913
Danna KJ, Sack GH Jr, Nathans D. 1973. Studies of simian virus 40 DNA. VII. A cleavage map of the SV40 genome. J Mol Biol 78: 363–376. 10.1016/0022-2836(73)90122-8
Hedgpeth J, Goodman HM, Boyer HW. 1972. DNA nucleotide sequence restricted by the RI endonuclease. Proc Natl Acad Sci 69: 3448–3452. 10.1073/pnas.69.11.3448
Hershfield V, Boyer HW, Yanofsky C, Lovett MA, Helinski DR. 1974. Plasmid ColEl as a molecular vehicle for cloning and amplification of DNA. Proc Natl Acad Sci 71: 3455–3459. 10.1073/pnas.71.9.3455
Hutchison CA III, Newbold JE, Potter SS, Edgell MH. 1974. Maternal inheritance of mammalian mitochondrial DNA. Nature 251: 536–538. 10.1038/251536a0
Jackson DA, Symons RH, Berg P. 1972. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc Natl Acad Sci 69: 2904–2909. 10.1073/pnas.69.10.2904
Judson HF. 1979. The eighth day of creation. The makers of the revolution in biology, a Touchstone book. Simon and Schuster, New York.
Kauc L, Piekarowicz A. 1978. Purification and properties of a new restriction endonuclease from Haemophilus influenzae Rf. Eur J Biochem/FEBS 92: 417–426. 10.1111/j.1432-1033.1978.tb12762.x
Kelly TJ Jr, Smith HO. 1970. A restriction enzyme from Hemophilus influenzae. II. J Mol Biol 51: 393–409. 10.1016/0022-2836(70)90150-6
Kleid D, Humayun Z, Jeffrey A, Ptashne M. 1976. Novel properties of a restriction endonuclease isolated from Haemophilus parahaemolyticus. Proc Natl Acad Sci 73: 293–297. 10.1073/pnas.73.2.293
Lai CJ, Nathans D. 1974. Deletion mutants of simian virus 40 generated by enzymatic excision of DNA segments from the viral genome. J Mol Biol 89: 179–193. 10.1016/0022-2836(74)90169-7
Lobban PE, Kaiser AD. 1973. Enzymatic end-to end joining of DNA molecules. J Mol Biol 78: 453–471. 10.1016/0022-2836(73)90468-3
Maxam AM, Gilbert W. 1977. A new method for sequencing DNA. Proc Natl Acad Sci 74: 560–564. 10.1073/pnas.74.2.560
Meselson M, Yuan R. 1968. DNA restriction enzyme from E. coli. Nature 217: 1110–1114. 10.1038/2171110a0
Morrow JF, Cohen SN, Chang AC, Boyer HW, Goodman HM, Helling RB. 1974. Replication and transcription of eukaryotic DNA in Escherichia coli. Proc Natl Acad Sci 71: 1743–1747. 10.1073/pnas.71.5.1743
Mowbray SL, Landy A. 1974. Generation of specific repeated fragments of eukaryote DNA. Proc Natl Acad Sci 71: 1920–1924. 10.1073/pnas.71.5.1920
Murray NE, Murray K. 1974. Manipulation of restriction targets in phage lambda to form receptor chromosomes for DNA fragments. Nature 251: 476–481. 10.1038/251476a0
Nathans D, Smith HO. 1975. Restriction endonucleases in the analysis and restructuring of DNA molecules. Ann Rev Biochem 44: 273–293. 10.1146/annurev.bi.44.070175.001421
Nathans D, Adler SP, Brockman WW, Danna KJ, Lee TN, Sack GH Jr, . 1974. Use of restriction endonucleases in analyzing the genome of simian virus 40. Fed Proc 33: 1135–1138.
Raleigh EA, Murray NE, Revel H, Blumenthal RM, Westaway D, Reith AD, Rigby PW, Elhai J, Hanahan D. 1988. McrA and McrB restriction phenotypes of some E. coli strains and implications for gene cloning. Nucleic Acids Res 16: 1563–1575. 10.1093/nar/16.4.1563
Roberts RJ. 1976. Restriction endonucleases. CRC Crit Rev Biochem 4: 123–164. 10.3109/10409237609105456
Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DT, Dybvig K, et al. 2003. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31: 1805–1812. 10.1093/nar/gkg274
Salser WA. 1974. DNA sequencing techniques. Ann Rev Biochem 43: 923–965. 10.1146/annurev.bi.43.070174.004423
Sanger F, Coulson AR. 1975. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 94: 441–448. 10.1016/0022-2836(75)90213-2
Sharp PA, Sugden B, Sambrook J. 1973. Detection of two restriction endonuclease activities in Haemophilus parainfluenzae using analytical agarose–ethidium bromide electrophoresis. Biochemistry 12: 3055–3063. 10.1021/bi00740a018
Smith HO, Nathans D. 1973. Letter: A suggested nomenclature for bacterial host modification and restriction systems and their enzymes. J Mol Biol 81: 419–423. 10.1016/0022-2836(73)90152-6
Smith HO, Wilcox KW. 1970. A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J Mol Biol 51: 379–391. 10.1016/0022-2836(70)90149-X
Southern EM. 1975a. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98: 503–517. 10.1016/S0022-2836(75)80083-0
Southern EM. 1975b. Long range periodicities in mouse satellite DNA. J Mol Biol 94: 51–69. 10.1016/0022-2836(75)90404-0
Takano T, Watanabe T, Fukasawa T. 1966. Specific inactivation of infectious lambda DNA by sonicates of restrictive bacteria with R factors. Biochem Biophys Res Commun 25: 192–198. 10.1016/0006-291X(66)90579-1
Takano T, Watanabe T, Fukasawa T. 1968. Mechanism of host-controlled restriction of bacteriophage lambda by R factors in Escherichia coli K12. Virology 34: 290–302. 10.1016/0042-6822(68)90239-0
Watanabe T, Nishida H, Ogata C, Arai T, Sato S. 1964. Episome-mediated transfer of drug resistance in enterobacteriaceae. VII. Two types of naturally occurring R factors. J Bacteriol 88: 716–726.
Watanabe T, Takano T, Arai T, Nishida H, Sato S. 1966. Episome-mediated transfer of drug resistance in enterobacteriaceae X. Restriction and modification of phages by fi R factors. J Bacteriol 92: 477–486.
Watson JD, Crick FH. 1953. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature 171: 737–738. 10.1038/171737a0
Watson JD, Tooze J. 1981. The DNA story: a documentary history of gene cloning. WH Freeman, San Francisco.
Yoshimori R, Roulland-Dussoix D, Boyer HW. 1972. R factor-controlled restriction and modification of deoxyribonucleic acid: Restriction mutants. J Bacteriol 112: 1275–1279.
WWW RESOURCES
http://library.cshl.edu/Meetings/restriction-enzymes/v-Bickle.php Bickle, T. 2013. Variations on a theme: the families of restriction/modification enzymes.
http://library.cshl.edu/Meetings/restriction-enzymes/v-Boyer.php Boyer, H. 2013. The discovery of EcoRI and its uses in recombinant DNA.
http://library.cshl.edu/Meetings/restriction-enzymes/v-Smith.php Smith, H. 2013. Discovery of the first Type II restriction enzyme and its aftermath.
http://rebase.neb.com/rebase/rebase.html The Restriction Enzyme Database.
1 http://library.cshl.edu/Meetings/restriction-enzymes/v-Smith.php. 2013.
