Restriction Enzymes: A History
By Wil A.M. Loenen, Leiden University Medical Center
April 2019 · 346 pages, illustrated (38 color and 26 B&W)
ISBN 978-1-621821-05-2
<< Appendix A — References >>
Appendix B
Chapter doi:10.1101/restrictionenzymes_AppB
Modern-Day Applications of Restriction Enzymes
In this book, the focus has been on the history of the development of the four types of REases with respect to their genetics, structure, and function, both in vivo and in vitro. Initially Type II REases such as EcoRI, HindIII, BamHI, and PstI have been used for genetic engineering of phage and plasmid vectors because of the presence or absence of their recognition sites in these phages and plasmids. These vectors have been extensively used for cloning, subcloning, DNA mapping, synthesis of large genetic scaffolds, and the study of chromatin structures and dynamics. In this appendix, S. Hong Chan discusses modern day applications of REases and nicking endonucleases (NEases) in molecular biology. Some of these REases (e.g., EcoP15I, FokI, MmeI, and NotI) have been discussed in detail in this book and are listed in Appendix 1 of Chapter 8. Many other REases can be found on the REBASE website (http://rebase.neb.com/rebase/rebase.html).
APPLICATIONS OF RESTRICTION ENDONUCLEASES: MOLECULAR CLONING AND BEYOND
Siu-Hong Chan
New England Biolabs, Inc.
From Molecular Cloning to Gene Assembly
Molecular Cloning
Restriction endonucleases and DNA ligases together facilitate a robust “cut and paste” workflow in which a defined DNA fragment (derived from cDNA or a cloned fragment) can be moved from one organism to another. The vehicles for cloning, plasmid vectors, were also created using this simple “cut and paste” methodology; original vectors, such as pSC101 and pBR322 (Cohen 2013), have gone through numerous generations of cutting and pasting with modules to become the backbone of many present-day vectors. For example, inserting promoters and origins of replication of eukaryotic viruses into these bacteriophage-derived plasmids has generated shuttle vectors, which are functional in both prokaryotic and eukaryotic cells. This cloning workflow, joined by DNA amplification technologies, such as PCR and RT-PCR, has facilitated the study of the molecular mechanisms of life (see Fig. 1).
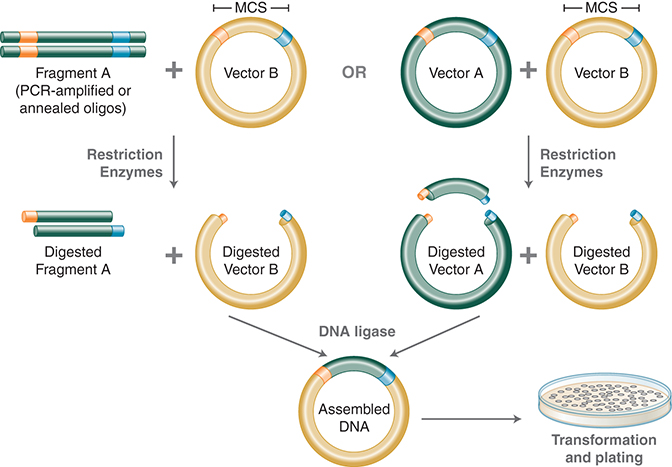
FIGURE 1. Traditional cloning workflow. Using PCR, restriction sites are added to both ends of a dsDNA, which is then digested by the corresponding REases. The cleaved DNA can then be ligated to a plasmid vector cleaved by the same or compatible REases with T4 DNA ligase. DNA fragments can also be moved from one vector into another by digesting with REases and ligating to compatible ends of the target vector. (Courtesy of NEB.)
In vitro DNA Assembly Technologies
Synthetic biology is a rapidly growing field in which defined components are used to create biological systems with precise control over the processes involved for the study of biological processes and the creation of useful biological devices (Ellis et al. 2011). Novel technologies such as BioBrick and USER Enzyme emerged to facilitate the building of such biological systems. Recently, more robust approaches such as Golden Gate Assembly, NEBuilder HiFi DNA Assembly, and Gibson Assembly have been widely adopted by the synthetic biology community. These approaches allow for parallel and seamless assembly of multiple DNA fragments without resorting to nonstandard bases.
BioBrick
The BioBricks community sought to create thousands of standardized parts for quick gene assembly (Stephanopoulos 2012). The BioBricks framework, together with the annual International Genetically Engineered Machines (iGEM) competition (www.igem.org), has elicited great interest from university and high school students around the world and helped inspire a whole new generation of synthetic biology scientists. Based on the traditional REase-ligation methodology, however, BioBrick and its derivative methodologies (such as BglBricks; Anderson et al. 2010) introduce scar sequences at the junctions and require multiple cloning cycles to create a working biological system.
USER Enzyme
USER (Uracil-Specific Excision Reagent) Enzyme is one of the first scar-less cloning technologies. It exploits the action of uracil DNA glycosylase and a pyrimidine lyase at a uracil incorporated into the PCR products through the primers (Nour-Eldin et al. 2010). USER Enzyme can therefore generate 3′ overhangs of custom sequences. Annealing of complementary overhangs allows multiple pieces of DNA to join together simultaneously and in order.
Golden Gate Assembly
Golden Gate Assembly and its derivative methods (Engler et al. 2008; Sarrion-Perdigones et al. 2011) exploit the ability of Type IIS REases to cleave DNA outside of the recognition sequence. The inserts and the cloning vectors are designed to place the Type IIS recognition site distal to the cleavage site, such that the Type IIS REase can remove the recognition sequence from the assembly. The advantages of such arrangement are threefold: (1) The overhang sequence created is not dictated by the REase and therefore no scar sequence is introduced; (2) the fragment-specific sequence of the overhangs allows orderly assembly of multiple fragments simultaneously; and (3) as the restriction site is eliminated from the ligated product, digestion and ligation can be carried out simultaneously. The net result is the ordered and seamless assembly of DNA fragments in one reaction. The accuracy of the assembly is dependent on the length of the overhang sequences. Therefore, Type IIS REases that create four-base overhangs (such as BsaI/BsaI-HF v2, BsmBI, Esp3I, BbsI/BbsI-HF, and EarI) are preferred. The downside of these Type IIS REase-based methods is that the small number of overhanging bases can lead to misligation of fragments with similar overhang sequences (Engler et al. 2009). Research has identified ligase bias on mismatch ligation sites to help guide the design of ligation junctions for high-fidelity assembly (Potapov et al. 2018a,b). As in REase-based cloning, it is also necessary to verify that the Type IIS REase sites used are not present in the fragments. Nonetheless, Golden Gate Assembly is a robust technology that generates multiple site-directed mutations (Yan et al. 2012) and assembles multiple DNA fragments into large contigs (Scior et al. 2011; Werner et al. 2012). As open source methods and reagents have become increasingly available (see www.addgene.org), Golden Gate Assembly has been widely used in the construction of genetic circuits (Halleran et al. 2018; Kong et al. 2017) among other applications. (See Fig. 2.)
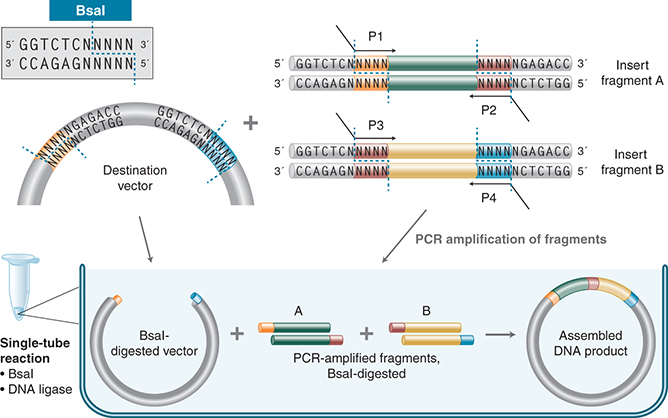
FIGURE 2. Golden Gate Assembly workflow. In its simplest form, Golden Gate Assembly requires a BsaI recognition site (GGTCTC) added to both ends of a dsDNA fragment distal to the cleavage site, such that the BsaI site is eliminated by digestion with BsaI. Upon cleavage, the overhanging sequences of the adjoining fragments anneal to each other. DNA ligase then seals the nicks to create a new covalently linked DNA molecule. Multiple pieces of DNA can be cleaved and ligated simultaneously. (Courtesy of NEB.)
Gibson Assembly, NEBuilder HiFi DNA Assembly, and Exonuclease-Based DNA Assembly Methods
Named after Daniel G. Gibson, Gibson Assembly is a robust exonuclease-based method to assembly DNA seamlessly and in sequence under isothermal conditions. It takes advantage of three complementary enzymatic activities to achieve a one-pot assembly of multiple pieces of DNA into a large contig: a 5′ exonuclease generates long 3′ overhangs, a polymerase fills in the gaps of the annealed single-stranded regions, and a DNA ligase seals the nicks of the annealed sequences (Gibson et al. 2009).
In addition to gene assembly, this and other commercially available assembly technologies (NEBuilder HiFi DNA Assembly from New England Biolabs and In-Fusion from Takara) can also be used for cloning; the assembly of a DNA insert with a linearized vector, followed by transformation, can be completed within a few hours. Other applications of these gene assembly methods include introduction of multiple mutations, assembly of plasmid vectors from chemically synthesized oligonucleotides, and creation of combinatorial libraries of genes and pathways. Reviews of DNA assembly methods are available in the literature (Merryman and Gibson 2012; Casini et al. 2015). (See Fig. 3.)
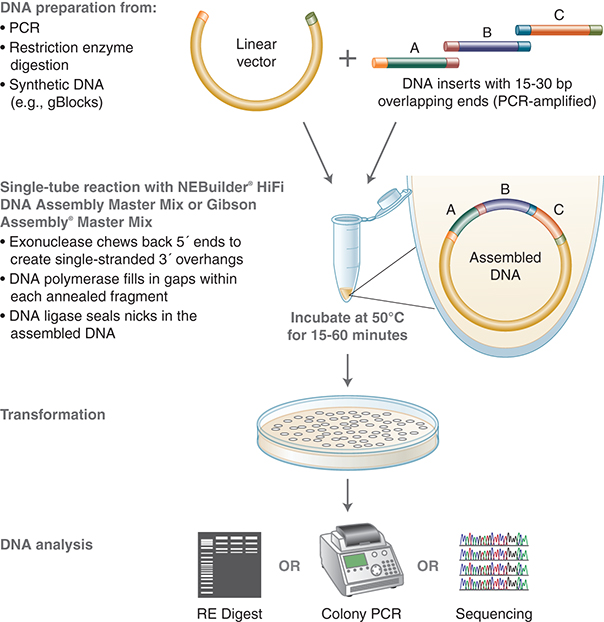
FIGURE 3. NEBuilder and Gibson Assembly workflow. NEBuilder and Gibson Assembly both employ three enzymatic activities in a single-tube reaction: 5′ exonuclease, the 3′-extension activity of a DNA polymerase, and DNA ligase activity. The 5′-exonuclease activity chews back the 5′-end sequences and exposes the complementary sequence for annealing. The polymerase activity then fills in the gaps on the annealed regions. A DNA ligase then seals the nick and covalently links the DNA fragments together. The overlapping sequence of adjoining fragments is much longer than those used in Golden Gate Assembly and therefore results in a higher percentage of correct assemblies. (Courtesy of NEB.)
From DNA Mapping to Chromatin Structural Dynamics
With only a handful of REases available in the early 1970s, Kathleen Danna in Daniel Nathan's group mapped the functional units of (simian virus) SV40 DNA (Danna and Nathans 1972) and thus commenced the era of eukaryotic gene mapping and comparative genomes. It has since evolved into sophisticated methodologies that allow the detection of single-nucleotide polymorphisms (SNPs) and insertions/deletions (indels) (Kudva et al. 2004), driving technologies that have facilitated genome-wide studies such as the mapping of epigenetic marks and chromatin structural dynamics and population-wide research such as population genomics of traits and genetic disorders.
Construction of DNA Libraries
Serial analysis of gene expression (SAGE) has been widely used to identify mutations in cancer research and study gene expression in transcriptome research. REases are key to creating ditags and concatamers in SAGE-type analyses. NlaIII is instrumental as an anchoring enzyme because of its unique property of recognizing a 4-bp sequence CATG and creating a 4-nt overhang of the same sequence. The use of Type IIS enzymes that cleave even further away from the recognition sequence as tagging enzyme allows the creation of longer ditags for higher information content of SAGE analyses: FokI and BsmFI in SAGE (Velculescu et al. 1995), MmeI in LongSAGE (Høgh and Nielsen 2008), and EcoP15I in SuperSAGE (Matsumura et al. 2012) and DeepSAGE (Nielsen 2008).
Although REases do not allow for the random fragmentation of DNA that most deep sequencing technologies require, they are used in target enrichment methodologies: hairpin adaptor ligation (Singh et al. 2011) and HaloPlex enrichment (Agilent). The long-reach REase AcuI and USER Enzyme were used to insert tags into sample DNA, which was then amplified using rolling circle amplification to form long single-stranded DNA “nanoballs” that served as template in a high-density ChIP-based sequencing by ligation methodology developed (Drmanac et al. 2010). ApeKI is used to generate DNA library for a genotyping-by-sequencing technology for the study of sequence diversity of maize (Elshire et al. 2011).
Mapping Epigenetic Modifications
REases have an extraordinary ability to discriminate the methylation status of the target bases. This property has been exploited to map modified bases within a genomic context. Before the advent of deep sequencing technologies, a two-dimensional gel electrophoresis–based mapping technique called restriction landmark genome scanning (RLGS) used NotI (GC^GGCCGC), AscI (GG^CGCGCC), EagI (C^GGCCG), or BssHII (G^CGCGC) to interrogate changes in the methylation patterns of the genome during development of normal and cancer cells. Methylation-sensitive amplification polymorphism (MSAP) takes advantage of the differential sensitivity of MspI and HpaII toward the methylation status of the second C in the sequence CCGG to map m5C, hm5C, and 5-glucosyl hydroxymethylcytosine (Reyna-López et al. 1997; Davis and Vaisvila 2011; Mastan et al. 2012). The REases that recognize and cleave DNA at 5-mC or 5-hmC sites, such as MspJI, FspEI, and LpnPI, are also potential tools for high-throughput mapping of the cytosine epigenetic markers in complex genomes (Cohen-Karni et al. 2011; Wang et al. 2011).
Point-of-Care DNA Amplification and Detection Using Nicking Endonucleases
By generating sequence- and strand-specific nicks on dsDNA, nicking endonucleases (NEases) open the door to applications that cannot be achieved by REases. In the presence of a strand-displacing DNA polymerase such as Bst DNA polymerase, the 3′-hydroxyl end of the nicked site can be extended for hundreds of nucleosides. Because the NEase site is regenerated, repeated nicking–extension cycles result in amplification of specific single-strand segments of the sample DNA without the need for thermocycling. Nicking enzyme–based isothermal DNA amplification technologies such as rolling circle amplification, NESA, EXPAR, and related amplification schemes have been shown to be capable of detecting very low levels of DNA (Dawson et al. 2009; Murakami et al. 2009). Similar schemes have been incorporated into molecular beacon technologies to amplify the signal (Li et al. 2008). The implementation of these sample and/or signal amplification schemes can lead to simple but sensitive and specific methods for the detection of target DNA molecules at point of care (e.g., NEAR, SDA, and EXPAR). This procedure is amenable to multiplexing and can potentially achieve higher fidelity than PCR. The combined activity of NEases and Bst DNA polymerase have also been used to introduce site-specific fluorescent labels into long/chromosomal DNA in vitro for visualization (nanocoding) (Kounovsky-Shafer et al. 2017). A general review of NEases and their applications has been published (Chan et al. 2011), and an excellent review of NEase-based DNA amplification and detection technologies and their application in molecular diagnostics is also available (Niemz et al. 2011).
Study of the Spatial Structure of Chromatin
Chromosome conformation capture (3C)-based technologies, such as 3C, 4C, 5C, and Hi-C, have revealed the important role of spatial proximity of genomic regions in genome rearrangements in cancer cells and the regulation of gene expression in general (Grob and Cavalli 2018). The core of the technologies—namely, 3C—involves the reversible formaldehyde crosslinking of DNA to proteins in the vicinity. After crosslinking, a six-base cutting restriction enzyme, normally HindIII or EcoRI, is used to cleave the contiguous genomic DNA into smaller units cross-linked to proteins. These DNA fragments, putatively spatial neighbors organized by the cross-linked proteins, are then ligated in such a way that intramolecular ligations are favored. For Hi-C-based technologies, the HindIII-cleaved DNA is filled in with biotinylated dA before ligation to allow for enrichment of the neighboring DNA downstream of the process. After the protein cross-links are reversed, the ligated DNA can be subjected to arrays of manipulations for loci-focused analysis (3C, 4C, or 5C) or deep-sequencing (Hi-C). Higher resolution of interacting regions can be achieved by using more frequent cutting restriction enzymes such as DpnII, MboI, and Sau3AI (four-base cutters) (Belaghzal et al. 2017).
Mapping of Open Chromatin Regions
The mammalian genome is largely packaged into chromatin consisting primarily of DNA and histones. Chromatin undergoes remodeling events that include switching between closed and open conformations to provide access to regulatory factors such as transcription factors. Hence, open chromatin profiling can provide information on the active regions of the genome under specific conditions. Tn5 transposase and DNase I–based sequencing methods (ATAC-seq and DNase-Seq, respectively) have been applied to map opened chromatin regions by virtue of the accessibility of the opened regions (Song and Crawford 2010; Buenrostro et al. 2013). Recently, a frequent nicking enzyme Nt.CviPII (recognition sequence = CCD, D being A, G, or T) has been used to establish the NicE-seq (nicking enzyme–assisted sequencing) method for open chromatin profiling at single-nucleotide resolution (Ponnaluri et al. 2017). In addition to being applicable to both native and formaldehyde-fixed cells, NicE-Seq has been shown to require a lower sequencing burden than DNase hypersensitive and ATAC-seq sites.
Genome Editing
At the infancy of genome editing, REases and homing endonucleases were the only available tools for creating double-strand breaks at nonspecific locations of the genome of higher organisms for transgenesis (Ishibashi et al. 2012a,b). In the 2000Ts, engineered enzymes such as zinc-finger nucleases (ZFNs) and transcription activator–like effector nucleases (TALENs) were developed and allowed genome editing operations, such as gene knockout and knock-in. The challenge, however, is the selection and screening of appropriate cleavage sites and the engineering of the enzyme for specific target sites.
In 2012, two seminal papers describing the adaptation of the bacterial CRISPR/CRISPR-associated systems (Cas) as RNA-guided DNA endonucleases (Gasiunas et al. 2012; Jinek et al. 2012) set off a new era of genome editing. These CRISPR/Cas systems can be adapted to use a single-guide RNA to direct the endonuclease activity to a target site within a complex genomic context both in vitro and in vivo. It is relatively simple to design and screen for appropriate guide RNA sequences and there is no need to engineer the enzyme. The simplicity and elegance of the CRIPSR–Cas systems has democratized genome editing, gene expression manipulation (by using catalytically inactive CRISPR–Cas proteins), and even RNA targeting (e.g., the Cas13 systems [Abudayyeh et al. 2017] and the prokaryotic Argonaute proteins [Dayeh et al. 2018]). The application of the CRISPR–Cas systems has been extensively reviewed (Nuñez et al. 2016; Huang et al. 2018; Knott and Doudna 2018). Comparative reviews of the three major genome editing methods are also available in the literature (Guha and Edgell 2017; Jaganathan et al. 2018; Yang and Wu 2018).
Looking Forward
Although restriction endonucleases have been one of the major forces that transformed molecular biology in the past decades, novel technologies such as Gibson Assembly, NEBuilder HiFi DNA Assembly, and Golden Gate Assembly continue to extend our ability to create new DNA molecules in vitro and CRISPR–Cas to edit and manipulate genomes in vivo. Restriction endonucleases and nicking endonucleases, meanwhile, have found new applications beyond molecular cloning in the age of deep sequencing and molecular diagnostics; genome-wide mapping of epigenetic marks, chromatin structural dynamics, and isothermal amplification and detection of genetic markers are all exciting and invaluable tools in the study of the molecular biology of life. As new technologies and new tools emerge, these highly sequence-specific endonucleases may find even more unique and exciting applications that help us understand the molecular mechanisms of life.
REFERENCES
Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al. 2017. RNA targeting with CRISPR-Cas13. Nature 550: 280–284.10.1038/nature24049
Anderson JC, Dueber JE, Leguia M, Wu GC, Goler JA, Arkin AP, Keasling JD. 2010. BglBricks: a flexible standard for biological part assembly. J Biol Eng 4: 1.10.1186/1754-1611-4-1
Belaghzal H, Dekker J, Gibcus JH. 2017. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 123: 56–65.10.1016/j.ymeth.2017.04.004
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218.10.1038/nmeth.2688
Casini A, Storch M, Baldwin GS, Ellis T. 2015. Bricks and blueprints: methods and standards for DNA assembly. Nat Rev Mol Cell Biol 16: 568–576.10.1038/nrm4014
Chan S-H, Stoddard BL, Xu S-Y. 2011. Natural and engineered nicking endonucleases—from cleavage mechanism to engineering of strand-specificity. Nucleic Acids Res 39: 1–18.10.1093/nar/gkq742
Cohen-Karni D, Xu D, Apone L, Fomenkov A, Sun Z, Davis PJ, Morey Kinney SR, Yamada-Mabuchi M, Xu S-y, Davis T, et al. 2011. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc Natl Acad Sci 108: 11040–11045.10.1073/pnas.1018448108
Cohen SN. 2013. DNA cloning: a personal view after 40 years. Proc Natl Acad Sci 110: 15521–15529.10.1073/pnas.1313397110
Danna KJ, Nathans D. 1972. Bidirectional replication of Simian Virus 40 DNA. Proc Natl Acad Sci 69: 3097–3100.10.1073/pnas.69.11.3097
Davis T, Vaisvila R. 2011. High sensitivity 5-hydroxymethylcytosine detection in Balb/C brain tissue. J Vis Exp 48: e2661.
Dawson ED, Taylor AW, Smagala JA, Rowlen KL. 2009. Molecular detection of Streptococcus pyogenes and Streptococcus dysgalactiae subsp. equisimilis. Mol Biotechnol 42: 117–127.10.1007/s12033-009-9143-2
Dayeh DM, Cantara WA, Kitzrow JP, Musier-Forsyth K, Nakanishi K. 2018. Argonaute-based programmable RNase as a tool for cleavage of highly-structured RNA. Nucleic Acids Res 46: e98.10.1093/nar/gky496
Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81.10.1126/science.1181498
Ellis T, Adie T, Baldwin GS. 2011. DNA assembly for synthetic biology: from parts to pathways and beyond. Integr Biol (Camb) 3: 109–118.10.1039/c0ib00070a
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. 2011. A Robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6: e19379.10.1371/journal.pone.0019379
Engler C, Gruetzner R, Kandzia R, Marillonnet S. 2009. Golden Gate shuffling: a one-Pot DNA shuffling method based on Type IIs restriction enzymes. PLoS One 4: e5553.10.1371/journal.pone.0005553
Engler C, Kandzia R, Marillonnet S. 2008. A one pot, one step, precision cloning method with high throughput capability. PLoS One 3: e3647.10.1371/journal.pone.0003647
Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci 109: E2579–E2586.10.1073/pnas.1208507109
Gibson DG, Yong L, Chuang R, Venter JC, Hutchison CA 3rd, . 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 44: 343–345.10.1038/nmeth.1318
Grob S, Cavalli G. 2018. Technical review: a hitchhiker's guide to chromosome conformation capture. Methods Mol Biol 1675: 233–246.10.1007/978-1-4939-7318-7_14
Guha TK, Edgell DR. 2017. Applications of alternative nucleases in the age of CRISPR/Cas9. Int J Mol Sci 18: 2565.10.3390/ijms18122565
Halleran AD, Swaminathan A, Murray RM. 2018. Single day construction of multigene circuits with 3G assembly. ACS Synth Biol 7: 1477–1480.10.1021/acssynbio.8b00060
Høgh AL, Nielsen KL. 2008. SAGE and LongSAGE. Methods Mol Biol 387: 3–24.10.1007/978-1-59745-454-4_1
Huang C-H, Lee K-C, Doudna JA. 2018. Applications of CRISPR–Cas enzymes in cancer therapeutics and detection. Trends Cancer 4: 499–512.10.1016/j.trecan.2018.05.006
Ishibashi S, Kroll KL, Amaya E. 2012a. Generating transgenic frog embryos by restriction enzyme mediated integration (REMI). In Methods in molecular biology (Clifton, N.J.), Vol. 917, pp. 185–203. Springer, New York.
Ishibashi S, Love NR, Amaya E. 2012b. A simple method of transgenesis using I-Sce I meganuclease in Xenopus. In Methods in molecular biology (Clifton, N.J.), Vol. 917, pp. 205–218. Springer, New York.
Jaganathan D, Ramasamy K, Sellamuthu G, Jayabalan S, Venkataraman G. 2018. CRISPR for crop improvement: an update review. Front Plant Sci 9: 985.10.3389/fpls.2018.00985
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821.10.1126/science.1225829
Knott GJ, Doudna JA. 2018. CRISPR–Cas guides the future of genetic engineering. Science 361: 866–869.10.1126/science.aat5011
Kong DS, Thorsen TA, Babb J, Wick ST, Gam JJ, Weiss R, Carr PA. 2017. Open-source, community-driven microfluidics with Metafluidics. Nat Biotechnol 35: 523–529.10.1038/nbt.3873
Kounovsky-Shafer KL, Hernandez-Ortiz JP, Potamousis K, Tsvid G, Place M, Ravindran P, Jo K, Zhou S, Odijk T, de Pablo JJ, et al. 2017. Electrostatic confinement and manipulation of DNA molecules for genome analysis. Proc Natl Acad Sci 114: 13400–13405.10.1073/pnas.1711069114
Kudva IT, Griffin RW, Murray M, John M, Perna NT, Barrett TJ, Calderwood SB. 2004. Insertions, deletions, and single-nucleotide polymorphisms at rare restriction enzyme sites enhance discriminatory power of polymorphic amplified typing sequences, a novel strain typing system for Escherichia coli O157:H7. J Clin Microbiol 42: 2388–2397.10.1128/JCM.42.6.2388-2397.2004
Li JJ, Chu Y, Lee BY-H, Xie XS. 2008. Enzymatic signal amplification of molecular beacons for sensitive DNA detection. Nucleic Acids Res 36: e36.10.1093/nar/gkn033
Mastan SG, Rathore MS, Bhatt VD, Yadav P, Chikara J. 2012. Assessment of changes in DNA methylation by methylation-sensitive amplification polymorphism in Jatropha curcas L. subjected to salinity stress. Gene 508: 125–129.10.1016/j.gene.2012.07.063
Matsumura H, Urasaki N, Yoshida K, Krüger DH, Kahl G, Terauchi R. 2012. SuperSAGE: powerful serial analysis of gene expression. In Methods in molecular biology (Clifton, N.J.), Vol. 883, pp. 1–17. Springer, New York.
Merryman C, Gibson DG. 2012. Methods and applications for assembling large DNA constructs. Metab Eng 14: 196–204.10.1016/j.ymben.2012.02.005
Murakami T, Sumaoka J, Komiyama M. 2009. Sensitive isothermal detection of nucleic-acid sequence by primer generation-rolling circle amplification. Nucleic Acids Res 37: e19.10.1093/nar/gkn1014
Nielsen KL. 2008. DeepSAGE: higher sensitivity and multiplexing of samples using a simpler experimental protocol. Methods Mol Biol 387: 81–94.10.1007/978-1-59745-454-4_6
Niemz A, Ferguson TM, Boyle DS. 2011. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol 29: 240–250.10.1016/j.tibtech.2011.01.007
Nour-Eldin HH, Geu-Flores F, Halkier BA. 2010. USER cloning and USER fusion: the ideal cloning techniques for small and big laboratories. Methods Mol Biol 643: 185–200.10.1007/978-1-60761-723-5_13
Nuñez JK, Harrington LB, Doudna JA. 2016. Chemical and piophysical modulation of Cas9 for tunable genome engineering. ACS Chem Biol 11: 681–688.10.1021/acschembio.5b01019
Ponnaluri VKC, Zhang G, Estève P-O, Spracklin G, Sian S, Xu S-Y, Benoukraf T, Pradhan S. 2017. NicE-seq: high resolution open chromatin profiling. Genome Biol 18: 122.10.1186/s13059-017-1247-6
Potapov V, Ong JL, Kucera RB, Langhorst BW, Bilotti K, Pryor JM, Cantor EJ, Canton B, Knight TF, Evans TC, et al. 2018a. Comprehensive profiling of four base overhang ligation fidelity by T4 DNA Ligase and application to DNA assembly. ACS Synth Biol 7: 2665–2674.10.1021/acssynbio.8b00333
Potapov V, Ong JL, Langhorst BW, Bilotti K, Cahoon D, Canton B, Knight TF, Evans TC, Lohman GJS. 2018b. A single-molecule sequencing assay for the comprehensive profiling of T4 DNA ligase fidelity and bias during DNA end-joining. Nucleic Acids Res 46: e79.10.1093/nar/gky303
Reyna-López GE, Simpson J, Ruiz-Herrera J. 1997. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol Gen Genet 253: 703–710.10.1007/s004380050374
Sarrion-Perdigones A, Falconi EE, Zandalinas SI, Juárez P, Fernández-del-Carmen A, Granell A, Orzaez D. 2011. GoldenBraid: an iterative cloning system for standardized assembly of reusable genetic modules. PLoS One 6: e21622.10.1371/journal.pone.0021622
Scior A, Preissler S, Koch M, Deuerling E. 2011. Directed PCR-free engineering of highly repetitive DNA sequences. BMC Biotechnol 11: 87.10.1186/1472-6750-11-87
Singh P, Nayak R, Kwon YM. 2011. Target-enrichment through amplification of hairpin-ligated universal targets for next-generation sequencing analysis. Methods Mol Biol 733: 267–278.10.1007/978-1-61779-089-8_19
Song L, Crawford GE. 2010. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc 2010: pdb.prot5384.10.1101/pdb.prot5384
Stephanopoulos G. 2012. Synthetic biology and metabolic engineering. ACS Synth Biol 1: 514–525.10.1021/sb300094q
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. 1995. Serial analysis of gene expression. Science 270: 484–487.10.1126/science.270.5235.484
Wang H, Guan S, Quimby A, Cohen-Karni D, Pradhan S, Wilson G, Roberts RJ, Zhu Z, Zheng Y. 2011. Comparative characterization of the PvuRts1I family of restriction enzymes and their application in mapping genomic 5-hydroxymethylcytosine. Nucleic Acids Res 39: 9294–9305.10.1093/nar/gkr607
Werner S, Engler C, Weber E, Gruetzner R, Marillonnet S. 2012. Fast track assembly of multigene constructs using Golden Gate cloning and the MoClo system. Bioeng Bugs 3: 38–43.
Yan P, Gao X, Shen W, Zhou P, Duan J. 2012. Parallel assembly for multiple site-directed mutagenesis of plasmids. Anal Biochem 430: 65–67.10.1016/j.ab.2012.07.029
Yang H, Wu Z. 2018. Genome editing of pigs for agriculture and biomedicine. Front Genet 9: 360.10.3389/fgene.2018.00360
