Restriction Enzymes: A History
By Wil A.M. Loenen, Leiden University Medical Center
April 2019 · 346 pages, illustrated (38 color and 26 B&W)
ISBN 978-1-621821-05-2
Chapter 5
Chapter doi:10.1101/restrictionenzymes_5
INTRODUCTION
The discovery of EcoRI was one of the most important scientific events of the 1970s (Yoshimori 1971; Yoshimori et al. 1972). It opened the way to molecular cloning as well as fundamental research into a large family of endonucleases with an extraordinary high specificity and fidelity. Interest in the Type II REases focused worldwide on their role as reagents, although initially the number of commercially available REases was small (Fig. 1). In several laboratories, hopes were high that REases might be good proteins to study in order to understand their enzymatic properties in DNA–protein interactions. Would this lead to new insights into DNA sequence specificity of REases (and other proteins)? Was there a single mechanism for the discrimination of base pairs? And how did REases distinguish their recognition sequences from the high background of nonspecific sequences?
FIGURE 1. Miles Labs' restriction enzymes catalog, circa 1980. (Courtesy of Bayer.)
The period between 1972 and 1982 marked the discovery of many new Type II enzymes, but few Type I and Type III enzymes. EcoRV is currently one of the best-characterized Type II enzymes, but the first important publications on EcoRV date to 1984–1985 (Bougueleret et al. 1984; Bougueleret 1985; Bougueleret et al. 1985; Chapter 6). After Hind II and EcoRI, EcoRII was among the very first REases for which the DNA substrate site was identified (5′C/C[A/T]GG) (Bigger et al. 1973; Boyer et al. 1973). A decade later, EcoRII would prove to be an interesting enzyme with rather different properties from EcoRI (Chapter 6).
During the 1970s, research focused mainly on the biochemistry and sequence recognition of EcoRI and a handful of other enzymes using plasmid, phage, or artificial DNA as substrate. Degradation was initially measured as the loss of biological activity or change in viscosity (Smith and Wilcox 1970; Gromkova and Goodgal 1972; Middleton et al. 1972; Takanami 1973; Chapters 1–4), but agarose gels soon simplified measurements greatly (Aaij and Borst 1972; Sharp et al. 1973). In particular, the arrival of submerged slab gels was met with great enthusiasm, as mentioned in Chapter 4. Note that although officially the EcoRI proteins should be called R·EcoRI and M·EcoRI to denote the REase and MTase, respectively, by convention the R is dropped in the case of the REase.
TYPE II ENZYMES
New Specificities and Isoschizomers of Type II Enzymes
The major feature that distinguishes one REase from another is its specificity: Where and how does the enzyme cut? The initial approach used by Kelly and Smith in 1970 (Kelly and Smith 1970) could reveal blunt end and/or 5′ or 3′ extensions at, or near, the recognition site (Roychoudhury and Kossel 1971; Murray 1973; Roychoudhury et al. 1976). In the 1970s, thanks to the computer (Brown and Smith 1977) (and agarose gels), banding patterns of new enzymes could be compared with those obtained with REases with known recognition sites. Was it a novel sequence or an isoschizomer of a known enzyme (Fuchs et al. 1978; Gingeras et al. 1978)? Additional experiments using primed synthesis reactions would then be used to confirm those predictions (Brown and Smith 1977; Brown et al. 1980). Isoschizomers were REases from different bacterial species that recognized the same sequence (Roberts 1976). This did not necessarily mean the same cleavage site (e.g., SmaI cleaves CCC/GGG and XmaI C/CCGGG; Endow and Roberts 1977). Such enzymes were later named neoschizomers (Hamablet et al. 1989).
By 1982, 360 Type II enzymes had been recognized, with more than 20 different sequence patterns of 85 specificities (Modrich and Roberts 1982). This was only the beginning of a meteoric rise: By 1993 this figure was nearly 2400 REases with 188 specificities (Roberts and Halford 1993; Roberts and Macelis 1993).
Recognition sites could be palindromic (like EcoRI), but also asymmetric, with ambiguities, or with unspecified bases in between the specific bases. Isoschizomers proved useful for various reasons: Amounts of REases were often found to vary substantially from strain to strain. This made one enzyme more commercially attractive than another or, alternatively, more feasible for biochemical or structural analysis. Also, isoschizomers could be differentially sensitive to methylation—for example, MboI could not cleave Gm6ATC, whereas Sau3A cleaved Gm6ATC (but not GATm5C [Sussenbach et al. 1976; Gelinas et al. 1977]). As the Escherichia coli Dam MTase (EcoDam, usually called Dam) methylates adenine in GATC sequences, Sau3A (/GATC) was extensively used to generate overlapping partial digests of DNA isolated from E. coli for gene libraries using the BamHI (G/GATTC) site in lambda (Loenen and Brammar 1980). Similarly, HpaII and MspI both recognized CCGG (Garfin and Goodman 1974), but only MspI could cleave Cm5CGG. This allowed screening eukaryotic genomes for the presence or absence of m5C sequences that might be involved in gene control (Waalwijk and Flavell 1978; Bird et al. 1979). Of course, such isoschizomers were also of particular scientific interest: Would these enzymes share common folds or have a common evolutionary origin?
Biochemistry of Type II Systems
By 1982, the MWs of more than 30 REases and MTases were known (Modrich and Roberts 1982). The few purified REases apparently contained a single polypeptide that existed in solution often as one or more oligomers, with some exceptions (Modrich and Roberts 1982). Did the aggregation state of the protein relate to the reaction mechanism (Lee and Chirikjian 1979)? In the case of EcoRI, the answer appeared to be positive: In solution, EcoRI was a mixture of dimers and tetramers, the former being stable at catalytic concentrations (10−10 M) (Modrich and Zabel 1976). This and other data supported the notion that EcoRI interacted with the DNA recognition site as a homodimer.
Although the REases varied considerably in MW (range of ∼22–70 kDa), the MTases were all more similar in length (range of ∼30–40 kDa). Another striking, and surprising, difference between the REases and MTases was that the five MTases analyzed (M·EcoRI, M·HpaI, M·HpaII, M·RsuI, and Dam) appeared to be monomers (Marinus and Morris 1973; Rubin and Modrich 1977; Yoo and Agarwal 1980b; Gunthert et al. 1981). Did this mean that the REase and MTase interacted with the shared (“cognate”) DNA recognition site in a significantly different way (Rubin and Modrich 1977)? And thus did not share a common evolutionary origin, despite recognizing the same DNA sequence?
The DNA Sequence of the EcoRI Genes
In 1981, the first sequence of a Type II R-M system was published—that of EcoRI. This allowed an answer to the burning question about the relationship between the REase and MTase (Greene et al. 1981; Jack et al. 1981; Newman et al. 1981). The REase gene encoded a protein of 277 amino acids (MW = 31 kDa) and the MTase gene a protein of 326 amino acids (MW = 38 kDa) (Newman et al. 1981; Rubin et al. 1981). Homology at the DNA or protein level was minimal; further experiments and computer predictions also made the existence of common features at a higher structural level unlikely (Chou and Fasman 1978a,b; Greene et al. 1981; Newman et al. 1981). This suggested that the two proteins would interact with DNA in different ways, which fit in with EcoRI being dimeric and M·EcoRI monomeric. The crystal structure of EcoRI in complex with DNA was eagerly awaited (McClarin et al. 1986; Kim et al. 1990).
Purification of EcoRI
The mechanism of DNA cleavage by EcoRI was examined in detail. The enzyme required only Mg2+ and unmodified DNA. Purification and early studies on the mechanism of cleavage were published by the groups of Paul Modrich in Durham (USA) (Modrich and Zabel 1976; Modrich and Rubin 1977; Rubin and Modrich 1978; Modrich 1979, 1982; Jack et al. 1980, 1981, 1982; Lu et al. 1981; Newman et al. 1981; Rubin et al. 1981; Young et al. 1981; Modrich and Roberts 1982; Cheng et al. 1984; Kim et al. 1984), Stephen (Steve) Halford in Bristol (UK) (Halford et al. 1979; Halford 1980; Halford and Johnson 1980, 1983), and John Rosenberg in Pittsburgh (USA), where he was joined later by Patricia (Pat) Greene (Rosenberg et al. 1978, 1980, 1981; Greene et al. 1981; Rosenberg and Greene 1982). Paul Modrich was awarded a PhD on E. coli DNA ligase with Robert Lehman at Stanford and was a postdoc with Charles Richardson at Harvard. At Duke University Medical Center he published on EcoRI from 1976 onward and became the leading expert in the field of strand-directed DNA mismatch repair, for which work he was awarded the Nobel Prize in Chemistry in 2015 (with Tomas Lindahl and Aziz Sancar). Steve Halford had done a PhD and postdoc with enzymologist Herbert Frederick (Freddie) Gutfreund on classical enzymes (e.g., lysozyme) at Bristol University. He switched to REases because he was intrigued by the extraordinary specificity of these enzymes and would study this in great detail. He was lucky to receive help from Nigel Brown, one of the first few people who had actually done a restriction digest! Nigel had mapped φX174 in Fred Sanger's laboratory, a technique he had learned from Rich Roberts’ technician Phyllis Myers (Halford 2013). John Rosenberg had obtained a PhD with Alexander Rich at Massachusetts Institute of Technology and a postdoc at Caltech with Richard Dickerson, before moving to Pittsburgh to elucidate the crystal structure of the EcoRI–DNA complex.
Both Halford and Rosenberg recalled their struggle to obtain enough EcoRI enzyme for their studies (Halford 2013; Rosenberg 2013). Halford's protocol for the purification of EcoRI in 1978 was a herculean task: “An enzymologist starts with about 100 mg enzyme!” (Halford 2013). He needed 800 L of bacteria with the EcoRI plasmid (Yoshimori et al. 1972), a 400-L fermentor at Porton Down, a bathtub, a rowing oar for stirring in a sackful of DEAE-cellulose (to absorb the EcoRI enzyme), and an end product of 10 mL of enzyme at 30,000,000 units/mL.
Rosenberg was even more desperate: He needed grams of EcoRI! Fortunately, Marc Zabeau made a strain overproducing EcoRI (Botterman and Zabeau 1985). This strain was useless for biochemical studies, as it produced insoluble protein, but useful for work on the crystal structure (Rosenberg et al. 1978; McClarin et al. 1986). Initially Rosenberg used the “Dickerson” dodecamer as DNA substrate (Drew et al. 1981). Sadly, this EcoRI–dodecamer complex gave poor crystals, but technical reasons (“Serendipity or how 13 can be a lucky number”) led to the addition of an extra T at the 5′ side of the 12-mer to give 5′-TCGCGAATTCGCG-3′, and those crystals were much better (Rosenberg 2013).
For his restriction analysis, Halford used a set of mutant lambda phages, each with only one of the five EcoRI recognition sites (Halford 2013). These were made by Noreen Murray from Edinburgh University, one of the “architects of the recombinant DNA revolution” (Gann and Beggs 2014), who would study the biology of EcoK and relatives for decades to come (Murray 2000, 2002). Stuart (Stu) Linn met Noreen and her husband Kenneth (Ken) Murray in Stanford in the 1960s, where they started their work that would result in many important contributions to the field of molecular biology and cloning DNA (see, e.g., Brockes et al. 1972; Bigger et al. 1973; Murray et al. 1973, 1975; Murray and Murray 1974; Old et al. 1975). In these papers (and in two complete books [Hershey 1971; Hendrix et al. 1983]) phage lambda played a prominent role and led the author of this book into the field.
Noreen had made these lambda mutants by cycling phages alternatively on an E. coli host with and without the EcoRI plasmid (Yoshimori et al. 1972) (Fig. 2). This work not only led to the first lambda vector for gene libraries (Murray and Murray 1974), but it also allowed Steve to study the kinetics of the reaction at each individual site in the same DNA context. Although the titer of the phage would drop ∼10-fold per site, the efficiency with which each of these sites was cut varied considerably (Halford and Johnson 1980), in line with similar observations by Thomas and Davis (1975). Did this support the notion of involvement of sequences external to the recognition site?
DNA Cleavage by Type II Enzymes
The study of DNA cleavage reactions was facilitated by a convenient biochemical manipulation involving the retention of site-specific complexes of EcoRI with DNA on nitrocellulose filters in the absence of Mg2+ (which blocks DNA cleavage) (Modrich and Zabel 1976; Modrich 1979; Halford and Johnson 1980; Jack et al. 1980; Rosenberg et al. 1980). These complexes underwent rapid cleavage upon addition of Mg2+. Depending on enzyme concentration and substrate, the enzyme dimer cut either one or both strands at the same time with a kcat value of 1–4 double-strand cuts/min/dimer at 37°C (Modrich and Roberts 1982). The results suggested a symmetrical complex between the EcoRI homodimer and the palindromic recognition site.
Further support for the influence of DNA outside the recognition site (Greene et al. 1975; Thomas and Davis 1975) came from limited data on seven other Type II enzymes—BamHI, HindIII, SalI, ClaI, BspI, HpaI, and HpaII—and studies with short synthetic DNA duplexes (Modrich and Roberts 1982). For example, different isoschizomers revealed differential strand preference for DNA duplexes with the sequence 5′-GAACCGGAA-3′: HpaII cut the complementary bottom (pyrimidine-rich) strand of the duplex three times faster than the upper strand; MspI cleaved the upper strand two times as fast; and a third isoschizomer, MnoI, attacked both strands at an equal rate (Baumstark et al. 1979; Yoo and Agarwal 1980a).
Methyl Transfer by Type II MTases
The kinetics of five purified MTases were simple: A monomer would transfer one methyl group at a time to the recognition site (Modrich and Roberts 1982). In contrast to Dam, EcoK, and EcoB (Vovis et al. 1974), M·EcoRI showed no preference for hemimethylated DNA (Modrich and Roberts 1982). These MTases were a novelty at the time: They were the first examples of monomeric proteins that interacted with twofold symmetrical DNA sequences.
Fidelity of Type II Enzymes
By the early 1970s, a variety of sequence-specific proteins were known to be able to bind DNA molecules lacking recognition sites, albeit with reduced affinity (von Hippel and McGhee 1972). Were Type II enzymes also capable of binding DNA nonspecifically? If so, was this sequence-independent or limited to sites that differed from the true (“canonical”) site by only one base pair? In vivo such errors could generate dangerous double-strand breaks: These had to be infrequent in order for cells to survive. Yet another question: Was infidelity a common feature of both REase and MTase, and, if so, would they recognize the same additional site(s)?
On a different track, various experiments suggested that EcoRI could bind sequences other than its cognate GAATTC. Was this an artifact? This question was raised by the fact that some, but not all, batches of EcoRI enzyme nicked DNA sites that differed by only one base pair from the canonical site (Tikchonenko et al. 1978; Bishop 1979; Bishop and Davies 1980; Maxam and Gilbert 1980).
In addition to this suspected purification artifact in some enzyme batches, EcoRI possessed solution-dependent relaxed (*) activity (much feared by early makers of gene libraries). EcoRI* appeared to preferentially cut G/AATTC but also /AATT sites (Polisky et al. 1975), but we now know (from Linda Jen-Jacobson and others) that that it is almost certain to be because of cutting at sites 1 and not 2 bp different from the recognition sequence. It is just that, under “odd” conditions such as low salt+high pH or Mn2+ in place of Mg2+ or with dimethylsulfoxide (DMSO) added, the difference in rate between cognate sites and sites 1 bp different is smaller than the more than million-fold difference under ideal conditions. The rates at sites 2 bp different are usually too slow to measure under any conditions. Various other enzymes, including MTases, showed relaxed specificity because of altered assay conditions: high enzyme concentrations, variations in pH and ionic strength, replacement of Mg2+ by Mn2+, or addition of glycerol or DMSO (Modrich and Roberts 1982). von Hippel and colleagues (Woodbury et al. 1980) made a valiant effort to examine the sequences cleaved under EcoRI* conditions by exploiting the availability of the total DNA sequence of φX174, which lacks EcoRI sites (Sanger et al. 1978). Although this analysis suggested preference for some sequences, results were not clear-cut and the origin and nature of EcoRI* activity remained unresolved.
Recognition of RNA–DNA Hybrids and/or ssDNA by Type II Enzymes
Both REases and MTases recognized dsDNA: Could they also recognize RNA–DNA hybrids and/or single-strand DNA (ssDNA)? The answer appeared to be a tentative yes, maybe: EcoRI, HaeIII, HhaI, HindII, and MspI cleaved RNA–DNA hybrids (prepared by reverse transcription) in a sequence-specific way (Molloy and Symons 1980). Experiments with denatured salmon sperm or T7 DNA indicated methylation by M·HaeII, M·HaeIII, and Dam (but not M·EcoRI or M·HpaI), albeit at reduced rates (Modrich and Roberts 1982). Likewise, some REases opened up single-stranded circular DNA of phages fl, M13, and φX174, again at reduced rates (Blakesley and Wells 1975; Horiuchi and Zinder 1975; Godson and Roberts 1976). Did this restriction or modification indeed occur at ssDNA, or did this happen within regions of (transient) secondary structure? Studies on HaeII, HpaI (Blakesley et al. 1977), and MspI (Yoo and Agarwal 1980a) led to two different models: In the case of the former (Blakesley et al. 1977), REase recognition was purported to occur only within regions of preformed secondary structure, whereas in the latter case, MspI would be able to form the duplex itself (Yoo and Agarwal 1980a). In contrast to these enzymes, EcoRI and HpaII clearly cut only the duplex form of their recognition sites (Greene et al. 1975; Baumstark et al. 1979). As the oligonucleotides used for HpaII and MspI were identical, apparently these enzymes differed in their interaction with their common recognition sequence (CCGG).
Concluding, neither in the experiments with RNA–DNA hybrids nor in those with ssDNA was evidence sufficient to exclude methylation or restriction occurring in (transient) duplex regions. Three decades later it was shown that a handful of REases (6/223 analyzed) were able to cut RNA–DNA hybrids—for example, Sau3AI can cut 80% of the DNA strand of an RNA–DNA hybrid (http://rebase.neb.com/rebase/rebase.html; Murray et al. 2010), and MspI can cut 5% of the DNA strand of a DNA–RNA duplex (http://rebase.neb.com/rebase/rebase.html). Such data suggest additional roles for some REases in vivo.
Specific versus Nonspecific Enzyme–DNA Interactions by Type II Enzymes
The above experiments suggested differential interaction of different REases and MTases with their recognition sequences. But at what stage did sequence specificity occur? Did the enzyme find the target site and bind? Or was the interaction weak and unstable until the moment of restriction or modification itself (i.e., at the level of catalyzing the phosphodiester bond by the REase) or by transferring the methyl group from SAM to DNA by the MTase? What were the contacts between protein and DNA: the bases and/or the backbone? Did the enzyme bind the major groove or the minor groove of the DNA?
The filter-binding experiments demonstrated that the EcoRI dimer interacted with the GAATTC sequence, with an apparent equilibrium dissociation constant for specific complexes between EcoRI and DNA with a single EcoRI site of ∼10−9–10−11 M (DNA of pBR322 or one of the lambda mutants used by Halford) (Modrich and Roberts 1982). In contrast, the nonspecific binding constant was in the micromolar range (Woodhead and Malcolm 1980). Like the lac repressor (Jovin 1976; von Hippel 1979), EcoRI also bound nonspecifically to a variety of polynucleotides in the presence of Mg2+ (Halford and Johnson 1980; Langowski et al. 1980), the nature of which was not understood (Modrich and Roberts 1982).
Specific complexes between EcoRI and DNA formed in the absence of Mg2+ were very stable with t1/2 ranging, surprisingly enough, from 16 to 140 min dependent on the chain length (range 6200 to 34 bp; Jack et al. 1980, 1982). This again implicated DNA sequences outside the recognition site (Jack et al. 1982). The rate of specific complex formation was also enhanced by DNA chain length, in line with preferential cleavage of longer DNAs in the presence of Mg2+ (Jack et al. 1982). The results were consistent with one-dimensional facilitated diffusion (Richter and Eigen 1974; Schranner and Richter 1978; Berg et al. 1981): that is, a sequence-specific protein would bind nonspecific sites and then diffuse randomly along the DNA helix until the recognition site was found (over a maximum distance of ∼1000 bp). Similar effects of DNA chain length on the kinetics of the lac repressor were noted (Winter et al. 1981).
Despite the ability of EcoRI to form site-specific complexes in the absence of Mg2+, attempts to detect specific binding by M·EcoRI in the absence of SAM were not successful (Modrich 1979; Jack et al. 1980; Woodhead and Malcolm 1980; Modrich and Roberts 1982).
Interactions of the EcoRI Enzymes with DNA
The above sequences of events indicated that EcoRI would bind the DNA and then slide along in order to find its cognate site. But how did it make specific contacts with the DNA: the major groove, the minor groove, and/or the backbone? To approach this question, base analogs were employed as well as partial chemical alkylation and alkylation interference. These provided the first tentative evidence that DNA contacts in the major groove were important in sequence recognition, and that the DNA–EcoRI complex possessed elements of twofold symmetry (Kaplan and Nierlich 1975; Berkner and Folk 1977; Modrich and Rubin 1977; Berkner and Folk 1979; Lu et al. 1981; Modrich and Roberts 1982). With respect to backbone contacts, ethylation interference studies suggested that four phosphates in each DNA strand were important in specific DNA–enzyme complexes, indicative of interactions with backbone phosphates outside the recognition site (Lu et al. 1981).
Attempts to elucidate the DNA contacts made by M·EcoRI and DNA involved analysis of analog effects on the kinetics of methyl transfer. The results of these studies were surprising and in marked contrast to those obtained with the REase—for example, T4 DNA has a bulky ghm5C base in the major groove, which makes the DNA resistant to EcoRI cleavage but an excellent substrate for M·EcoRI (Berkner and Folk 1977; Modrich and Rubin 1977; Modrich and Roberts 1982). These results were striking and lend support to the conclusion that the two proteins not only interacted with their cognate sites in different ways but also used different mechanisms to discriminate a given base pair.
Other Type II R-M Systems
The only other Type II system examined by 1982 in some detail was HpaI (Mann and Smith 1979). The results resembled those of EcoRI (Mann and Smith 1979). In an interesting experiment, a synthetic duplex oligonucleotide (5′-NxCCGGCCNx) with overlapping HpaII (CCGG) and HaeIII (GGCC) sites was used to examine the effects of m5C methylation on cleavage by the restriction enzymes (Mann and Smith 1979). Did M·HpaII modification affect HaeIII restriction and/or vice versa? Apparently they did: HaeIII cleaved 5′-GGCm5C as opposed to the normally modified site 5′-GGm5CC, which is resistant. Similarly, HpaII cleaved 5′-CCGG with m5C opposite the second G, but not 5′-CCGG.
Preliminary studies with other systems also supported the notion that different enzymes used different mechanisms to recognize a given base pair (Marchionni and Roufa 1978; Berkner and Folk 1979). This was further supported by the use of a random copolymer d(G,C) and analogs of this polynucleotide (Mann and Smith 1979), although other copolymer studies were inconclusive (Mann et al. 1978; Modrich and Roberts 1982).
TYPE I SYSTEMS
Genes and Proteins of Type I Systems
Although the hunt was on for new Type II enzymes, no extensive survey of bacteria was made for the presence of ATP-dependent REases. No techniques were available in the 1980s for the rapid identification of Type I sites but at that time sites of cleavage by Type II REases could easily be identified. Besides, unlike Type II, there was no commercial/technological driving force to find new Type I systems. During this decade, Noreen Murray in Edinburgh and Bob Yuan and Tom Bickle in Basel instigated much of the genetics and biochemistry. By 1982, all known Type I REases had been found in Enterobacteriaceae, with the exception of one enzyme in Haemophilus (HindI) (Gromkova and Goodgal 1976; Bickle 1982). These systems were carried by different strains of E. coli and Salmonella typhimurium, were mapped to the same location on the chromosome as EcoK and EcoB, and were allelic (Chapter 2; Boyer 1964; Arber and Wauters-Willems 1970; Glover 1970; Bullas and Colson 1975; Bachmann and Low 1980; Bullas et al. 1980). Two genes, hsdM and hsdS, were essential for modification, and a third gene, hsdR, was essential for restriction.
The hsdK locus of E. coli K12 was the first Type I system to be cloned (Borck et al. 1976). This allowed DNA sequence analysis of the smallest gene, hsdS (Sain and Murray 1980). By deletion analysis the gene order was shown to be hsdR, hsdM, hsdS, with the hsdM and hsdS genes transcribed from one promoter and the hsdR gene from a separate promoter in the same direction (Sain and Murray 1980).
What was the physical relationship between these systems? Using the novel Southern blot technique (Southern 1975), strong DNA cross-hybridization was detected with DNA probes derived from hsdM and hsdR of E. coli K12 and B; DNA derived from E. coli C did not have DNA sequences that hybridized to these probes, in line with the lack of R-M activity (Sain and Murray 1980; Murray et al. 1982). This killed the argument, often made at the time, that the Type I enzymes were “so complicated” that they must perform an additional, vital role in the cell. As E. coli C was perfectly viable, this was clearly not the case.
Antibodies raised against the purified HsdM and HsdR subunits of EcoK cross-reacted with purified HsdR and HsdM proteins of EcoB as expected (Murray et al. 1982). These antibodies also cross-reacted with HsdR and HsdM from three S. typhimurium strains (SB, SP, and SQ), but not with another E. coli strain, E. coli A (Arber and Wauters-Willems 1970). This would lead later to a subdivision into Type IA and IB.
Recognition Sequences of Type I Systems
As the Type I enzymes did not cut at their recognition sites, they were difficult to find. The analysis of mutant sequences was the solution to this problem. By the late 1970s the recognition sequences of EcoK and EcoB finally became known and were thought rather unusual: two specific sequences, a trimer and a tetramer, separated by a nonspecific sequence, and without any symmetry. The EcoB recognition sequence was 5′TGA(N8)TGCT (Lautenberger et al. 1978, 1979; Ravetch et al. 1978; Sommer and Schaller 1979), that of EcoK 5′-AAC(N6)GTGC (Kan et al. 1979). The adenine methylated in the EcoB sequence would be on the single A in the top strand and (most likely) the first A on the bottom strand (van Ormondt et al. 1973); in the case of EcoK the single A in the bottom strand was the obvious target for methylation. In the top strand, the second adenine was the site of methylation, as shown by Ineichen and Bickle: They cleverly used one of the EcoK sites in lambda DNA, where the trimeric part of the recognition sequence (AAC) overlaps with a HindII site (GTAAC) (Fig. 2 in Bickle 1982).
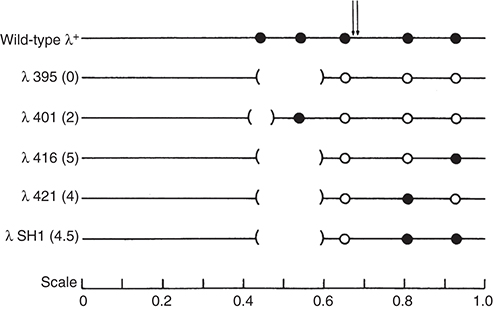
FIGURE 2. Bacteriophage lambda DNA contains five EcoRI sites. Lambda mutants lacking one or more EcoRI sites were useful to assay REase activity both in vivo and in vitro at individual sites. (Closed circle) EcoRI site, (open circle) mutant site, ( ) deletion removing one or two EcoRI sites. (Reprinted from Halford et al. 1980, with permission from Portland Press.)
Reaction Mechanisms of Type I Systems
Most of the data on the reaction scheme were derived from work on EcoK, but it likely applied to the other Type I enzymes (Bickle 1982). EcoK had at least three binding sites for its cofactor SAM and no detectable affinity for DNA in the absence of SAM. Addition of SAM led to a slow allosteric change, and this form could bind DNA (Hadi et al. 1975). The activated enzyme did not require free SAM in order to restrict and could bind nonspecifically to DNA (Yuan et al. 1975). Subsequently, the enzyme could bind tightly to specific sites and, in the absence of ATP, to both modified and unmodified sites. Such complexes were very stable (like EcoRI; see above): The t1/2 of a complex with a modified site was ∼6 min, and with an unmodified even as long as 22 min (Yuan et al. 1975).
Once firmly bound to the site, one of three things would happen: if the DNA was modified, the enzyme would be released from the DNA, as shown by both gel filtration and EM studies (Bickle et al. 1978). On heteroduplex DNA (one strand modified), methylation was efficient, stimulated by ATP (Vovis et al. 1974; Burckhardt et al. 1981; Bickle 1982). Such a scenario made sense (Bickle 1982): Newly replicated DNA must be fully methylated before the next round of replication to avoid double-strand breaks. It would be shown later that EcoK does not kill E. coli so easily (Chapter 7)! In the third event, that of unmodified DNA, surprisingly, the enzyme underwent a large conformational change: It remained bound to the recognition site, but generated large loops visible in the EM (Bickle et al. 1978; Rosamond et al. 1979). The enzyme would pump the DNA past itself and DNA cleavage would occur somewhere between 400 and 7000 bp away (Horiuchi and Zinder 1972; Adler and Nathans 1973; Bickle et al. 1978; Rosamond et al. 1979; Yuan et al. 1980a). Here, a difference was noted between EcoK and EcoB: Loops by EcoK were bidirectional, and those of EcoB were one-directional (and away from the trimeric part of the recognition sequence). No formal explanation has presented itself until the day of writing, but it is tacitly assumed that EcoK has two HsdR subunits, and EcoB only one during the restriction process.
Whatever the exact mechanism, the cleavage reaction itself was a two-step process: The DNA was nicked in one strand and then, seconds later, in the second strand. It was already noted that in the presence of excess DNA, only single-strand breaks were found, suggesting that two enzyme molecules were required for double-strand cleavage (Meselson and Yuan 1968; Roulland-Dussoix and Boyer 1969; Lautenberger and Linn 1972; Adler and Nathans 1973).
Several other curious and unexplained observations were made, the most puzzling one that of massive continued ATP hydrolysis after restriction, with hydrolysis of an estimated 10.000 ATP/min/enzyme molecule (see Bickle 1982 for further discussion).
TYPE III ENZYMES
By 1982, only three members had been identified. In addition to the system of Joe Bertani's phage P1, EcoP1, a second system was found on a plasmid in E. coli 15T− (EcoP15) and the third in Haemophilus influenzae Rf (HinfIII, probably chromosomal) (Arber and Dussoix 1962; Arber and Wauters-Willems 1970; Piekarowicz et al. 1974). EcoP1 and EcoP15 were allelic and highly homologous. Based on their behavior after methionine starvation in the presence of ethionine, they were initially grouped with Type II enzymes, but as their substrate requirements were more complex, they were designated Type III in 1978 (Kauc and Piekarowicz 1978). The EcoP1 enzyme was the first Type III enzyme to be purified (Haberman 1974).
Genetics of Type III Systems
The genetics of the P1 system initially suggested that EcoP1 required three genes for restriction (Scott 1970; Rosner 1973; Reiser 1975), but transposon mutagenesis indicated only two genes, mod ahead of res, which were transcribed in the same direction from their own promoter (Iida et al. 1983). Both EcoP1 and EcoP15 enzymes contained two subunits of ∼75 kDa (Mod) and 100 kDa (Res) (Heilmann et al. 1980; Hadi et al. 1983), which showed immunological cross-reactivity with each other (Fig. 5 in Bickle 1982).
DNA Recognition and Cleavage Sequences of Type III Systems
The recognition sequences were established for EcoP1 (5′AGACC), EcoP15 (5′CAGCAG), and HinfIII (5′CGAAT). Like EcoK and EcoB, methylation occurred on adenine (Brockes et al. 1972; Reiser 1975; Kauc and Piekarowicz 1978). It would remain a puzzle for quite some time and an interesting biological problem—that is, how the cell avoided cell death in the case of EcoP1 and EcoP15, as only the strand shown here can be modified (the opposite strand lacks adenine residues). All three enzymes cleaved the DNA ∼25–27 bp away to the right of the sequence shown, with two to three base single-strand 5′ extensions. It was estimated that this distance of ∼9 nm could easily be spanned by the enzyme without the enzyme having to move along the DNA. A likely model for the action of the enzyme was proposed: The modification subunit would recognize the specificity site and direct the restriction subunit to the site to be cleaved (Bickle 1982).
Reaction Mechanisms of Type III Systems
The reaction mechanisms of all three enzymes were rather similar: All three required ATP for cleavage, but ATP was not hydrolyzed in the process. SAM was not essential, although it stimulated the restriction process. In the presence of both cofactors, restriction and modification competed with each other. Working with these enzymes was complicated for various reasons, making the interpretation of results often difficult (see Bickle 1982 for discussion). SAM was the methyl donor for modification (Yuan and Reiser 1978). The enzyme was able to bind DNA in complex with SAM but also in the absence of cofactors, as seen in the EM (Yuan et al. 1980b). The Type III enzymes did not cleave or methylate each of their recognition sites with equal efficiency: A good methylation site was not necessarily a poor restriction site and vice versa (Yuan et al. 1980b).
In conclusion, a lot of mysterious aspects of the biology and reaction mechanisms of the Type III enzymes remained to be explained. Why were the enzymes inefficient nucleases in vitro but not in vivo? Why was only one strand of the recognition sequence modified (similar to the Type II REase MboII) (Bachi et al. 1979). How did the cell avoid restricting these sites? It left Tom Bickle with an “uncomfortable feeling that something basic” was missing from their in vitro systems (Bickle 1982). It would take decades to find answers to the many puzzling questions with respect to the Type III enzymes. To mention just two landmark papers in the history of the Type III enzymes: Tom Bickle and his coworkers resolved one of the mysterious aspects in 1992, when they showed that the Type III enzymes needed two inversely orientated sites for restriction (Meisel et al. 1992; see Chapter 6), whereas another major breakthrough would be published by Aneel Aggarwal and colleagues in 2015, when they published the structure of EcoP15I (Gupta et al. 2015; see Chapter 8).
